
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zinc diethyldithiocarbamate as a catalyst for synthesising biomedically-relevant thermogelling polyurethanes†
Jason Y. C.
Lim
 *a,
Qianyu
Lin
a,
Connie K.
Liu
ab,
Liangfeng
Guo
b,
Kun
Xue
a and
Xian Jun
Loh
*a
*a,
Qianyu
Lin
a,
Connie K.
Liu
ab,
Liangfeng
Guo
b,
Kun
Xue
a and
Xian Jun
Loh
*a
aInstitute of Materials Research and Engineering (IMRE), A*STAR (Agency for Science, Technology and Research), 2 Fusionopolis Way, Innovis, #08-03, Singapore 138634, Singapore. E-mail: lohxj@imre.a-star.edu.sg; jason_lim@imre.a-star.edu.sg
bInstitute of Chemical and Engineering Sciences (ICES), A*STAR (Agency for Science, Technology and Research), 1 Pesek Road, Jurong Island, Singapore 627833, Singapore
First published on 28th October 2020
Abstract
Zinc dithiocarbamates are traditionally highly-efficient catalysts for accelerating rubber vulcanization reactions, but have thus far not found substantial use in other forms of polymerisations. Herein, we demonstrate a new application of zinc diethyldithiocarbamate (ZDTC) for the synthesis of polyurethanes suitable for biomedical applications. Compared with other organozinc compounds such as zinc acetylacetonate and zinc neodecanoate, ZDTC showed exceptional catalytic performance, capable of forming high molecular weight polyurethanes, with the reaction tolerant to different organic solvents. The ZDTC-catalysed biocompatible amphiphilic polyurethanes comprising PEG and PPG blocks form thermo-responsive hydrogels which showed remarkably low sol-to-gel transition temperatures at surprisingly low concentrations in water. Microstructural analysis of the polyurethane aqueous solutions by small-angle X-ray scattering (SAXS) experiments revealed considerable supramolecular self-assembly of the polyurethanes even at 4 °C. Valuably, cell viability studies revealed that residual ZDTC catalysts present in unpurified polyurethane polymers exerted considerably lower cytotoxicities than those catalysed by tin(IV) catalysts such as dibutyltin dilaurate (DBTL). As demonstrated herein, ZDTC may be a possible alternative to highly-toxic organotin catalysts for the synthesis of polyurethanes, with further potential for other polymerization reactions reliant on organotin Lewis acidity such as polyester synthesis.
Introduction
Polyurethanes (PUs) are one of the most important classes of polymers, accounting for approximately 5% of global polymer production.1 Their popularity stems partially from the diverse range of readily-accessible monomers available, allowing PUs to be used in many applications such as foams, fibres and sealants. In recent years, the use of biocompatible building blocks such as poly(ethylene glycol) (PEG) and poly(propylene glycol) (PPG) has opened up new possibilities as biomaterials,2,3 including polyurethane thermoresponsive hydrogels. These thermogels, which usually comprise a hydrophilic component (e.g. PEG) and at least one other hydrophobic component (e.g. poly(propylene glycol) (PPG), polycaprolactone and polyhydroxybutyrate), can undergo sol–gel transition as the temperature of the solution is increased,4 driven by the dehydration of the hydrophobic component and hydrogen bonding between the urethane linkages. This property makes these gels especially valuable as they can gelate in situ in the body at physiological temperature, allowing their use as localised depots for the controlled release of drugs.5–16 Recently, PU thermogels have also been demonstrated to function as effective vitreous tamponades for vitreoretinal surgery that facilitate vitreous regeneration.17The most straightforward and ubiquitous method of producing PUs is via the polyaddition of diol and diisocyanate monomers in the presence of a catalyst. However, synthesis of many PUs still relyheavily on the use of organotin catalysts, such as dibutyltin dilaurate (DBTL),18,19 to achieve the desired polymer molecular weights and bulk material properties. Indeed, the use of organotin catalysts for the polyaddition of alcohols to isocyanates to form PUs is well-established due to their catalytic efficiency,18,20 as well as excellent tolerance to heat and oxidation. However, these compounds are highly toxic,3,21 and their stability makes them highly persistent in the environment as well.4 Furthermore, they are difficult to remove in the resulting polymers, necessitating prohibitive costs to do so,22,23 which is especially critical for PUs used in biomedical applications due to the catalyst's potential cytotoxicity. As a result, a number of studies have investigated the use of tin-free catalysts for PU synthesis from polyols and isocyanates, either with neat monomers or in solution. Organocatalysts such as cyclic guanidines,24 N-heterocyclic carbenes (NHCs),25 tertiary amines,26 and organic acids including diphenyl phosphate and triflic acid27 were shown to be effective, although some examples such as NHCs are inherently unstable and tertiary amines containing N,N-dimethyl groups are prone to aerobic oxidation.28 Metallic catalysts containing transition metals (Cr, Fe, Cu, Ti, Mn, Ru, Hf), lanthanides (La) and main group metals (Bi, Ga, In, Al) have also shown varying degrees of catalytic activity for PU synthesis,29,30 despite the known toxicity and adverse health effects resulting from some of these metals,31–35 and their environmental concerns.36–38 Moreover, many organometallic catalysts are highly susceptible to deactivation by the presence of moisture, necessitating scrupulously dry reaction conditions.39,40
Compared to many metals, zinc possesses relatively low toxicity41,42 and is an essential trace element for the growth and development of higher plants and animals, including humans.43 Zinc has featured prominently as catalysts to synthesise numerous classes of polymers owing to its Lewis acidity, including polyesters by lactone ring-opening polymerisation,44,45 polyethylene,46–48 polycarbonates by ring-opening copolymerisation of carbon dioxide with epoxides,49,50 have been highly promising for polyurethane synthesis30,51 and are known activators and accelerators of rubber vulcanisations.52 The last class of reactions prominently feature zinc dithiocarbamates.53 Mechanistic studies have revealed that these catalysts can accelerate the sulfuration of the rubber backbone, the formation of polysulfide crosslinks and inducing desulfhydration of polythiothiols by H2S formation.54 More recently, zinc diethyldithiocarbamate (ZDTC) was demonstrated to effectively catalyse inverse vulcanisation reactions.55 However, apart from vulcanisation reactions, the applications of zinc dithiocarbamates as catalysts for other classes of polymerisation reactions remain largely unexplored. In this manuscript, we report the use of ZDTC as a potent non-tin alternative to DBTL in the synthesis of biocompatible thermogelling PUs. We show that ZDTC is able to catalyse the synthesis of high molecular weight PUs using PEG- and PPG-diol macromonomers, which are able to form mechanically-robust hydrogels at low polymer concentrations in water. Furthermore, SAXS analysis of the polymer solutions at different temperatures confirmed their temperature-responsiveness at the microstructural level, showing contrasting polymer hierarchical self-assembly behaviour compared to their DBTL-catalysed counterparts in solution which were evident even at 4 °C. Cell viability assays confirmed the significantly lower toxicity of crude unpurified ZDTC-catalysed PUs compared with those by DBTL towards mouse fibroblast cells.
Material and methods
General: materials and characterisations
PEG with average Mn of 2050 g mol−1, PPG with average Mn of 2000 g mol−1, 1,6-hexamethylene diisocyanate (HMDI, 99%), dibutyltin dilaurate (DBTL, 95%), zinc diethyldithiocarmate (ZDTC, 97%), zinc acetylacetonate (Zn(acac)2), zinc neodecanoate, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), and bismuth(III) neodecanoate were purchased from Sigma-Aldrich. Anhydrous toluene was purchased from Tedia with moisture content quantified by Karl Fischer titrations to be approximately 40 ppm. N,N-Dimethylformamide (DMF), chlorobenzene, diethyl ether and hexane were purchased from Sigma Aldrich. All chemicals, reagents and solvents were used as received without further purification. DMEM (Dulbecco's Modified Eagle Medium), fetal bovine serum (FBS), sterile-filtered 1% (wt/v) penicillin–streptomycin, sterile 1× phosphate buffered saline, 0.25% trypsin–EDTA, and sodium dodecyl sulfate were purchased from Sigma-Aldrich. Haemocytometer was purchased from Bio-Rad. NIH3T3 mouse fibroblasts were purchased from American Type Culture Collection (ATCC). Cell Titer Blue reagent kit was purchased from Promega (Germany). 25 cm2 standard cell culture flasks were purchased from Greiner Bio-One. Nalgene™ Rapid-Flow™ Sterile Disposable Filter Units with PES membrane and 0.2 μm pore size is purchased from Thermofisher Scientific.Polyurethane synthesis
3.33 g of commercially-available PEG (Mn 2050) and 1.67 g of PPG (Mn 2000) were first dried by azeotropic distillation using anhydrous toluene (2 × 20 mL). Following complete solvent removal in vacuo for an hour at 110 °C, the polymers were then re-dissolved in anhydrous toluene (30 mL) at 110 °C. To this solution the catalyst (0.0067 mol equivalents with respect to each hydroxyl/isocyanate group present) was added, followed by HMDI (0.40 mL, to maintain a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 isocyanate:hydroxyl mole ratio) portionwise successively. After stirring at 110 °C for 24 h under an Argon atmosphere, 5 mL of ethanol was added to the reaction and stirred for a further 15 minutes to quench any unreacted isocyanate groups present. The polymer solution was then precipitated by pouring into vigorously-stirred diethyl ether (400 mL). For polymer reactions which were highly viscous, such as that catalysed by ZDTC, the reaction was first further diluted with toluene (20 mL) before precipitation. The suspension was filtered and the polymeric residue was blown dry using compressed air to afford the crude polymer as a white solid. To further remove unreacted low molecular oligomers, the polymers were purified by dialysis for 3 days in distilled water using dialysis tubing (MWCO 3500 Da). The dry polymer fibres were then obtained by freeze-drying the polymers for 7 days (purified yield 24–91%, depending on catalyst used). 1H NMR (CDCl3, 500 MHz): 4.88 (br. s, OCON
:1 isocyanate:hydroxyl mole ratio) portionwise successively. After stirring at 110 °C for 24 h under an Argon atmosphere, 5 mL of ethanol was added to the reaction and stirred for a further 15 minutes to quench any unreacted isocyanate groups present. The polymer solution was then precipitated by pouring into vigorously-stirred diethyl ether (400 mL). For polymer reactions which were highly viscous, such as that catalysed by ZDTC, the reaction was first further diluted with toluene (20 mL) before precipitation. The suspension was filtered and the polymeric residue was blown dry using compressed air to afford the crude polymer as a white solid. To further remove unreacted low molecular oligomers, the polymers were purified by dialysis for 3 days in distilled water using dialysis tubing (MWCO 3500 Da). The dry polymer fibres were then obtained by freeze-drying the polymers for 7 days (purified yield 24–91%, depending on catalyst used). 1H NMR (CDCl3, 500 MHz): 4.88 (br. s, OCON![[H with combining low line]](https://www.rsc.org/images/entities/i_char_0048_0332.gif) ), 4.19 (m, C2OCONH); 3.63 (br. s, OC2 of PEG); 3.53 (m, OC2 of PPG); 3.39 (m, CCH3 of PPG); 3.14 (m, NC2CH2CH2); 1.47 (m, NCH2C2CH2); 1.31 (m, NCH2CH2C2); 1.13 (m, CHC3 of PPG) (see Fig. 1A for full spectra).
), 4.19 (m, C2OCONH); 3.63 (br. s, OC2 of PEG); 3.53 (m, OC2 of PPG); 3.39 (m, CCH3 of PPG); 3.14 (m, NC2CH2CH2); 1.47 (m, NCH2C2CH2); 1.31 (m, NCH2CH2C2); 1.13 (m, CHC3 of PPG) (see Fig. 1A for full spectra).
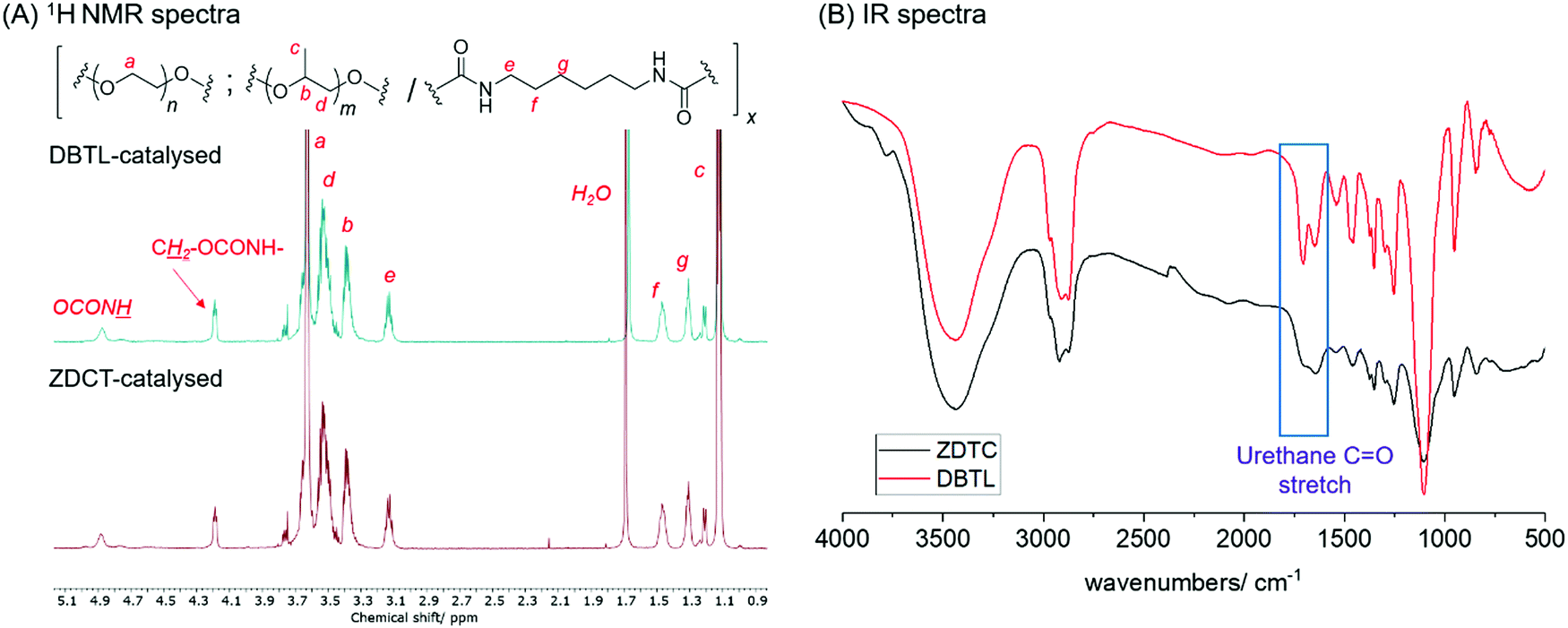 | ||
| Fig. 1 Comparison of the PUs synthesized using DBTL and ZDTC with (A) 1H NMR (500 MHz, CDCl3) and (B) FTIR spectroscopy. | ||
The mole percentage of each component (PEG2050, PPG2000 and HMDI) were calculated by integrating the multiplets at 1.08–1.14 ppm for PPG, 3.10–3.15 ppm for HMDI and 1.08–1.14 ppm for PEG.
Polyurethane molecular characterisation
Nuclear magnetic resonance (NMR) spectra were recorded at room temperature on a JEOL ECA 500 MHz NMR spectrometer operating at 500 MHz, with the samples dissolved in CDCl3 or d7-toluene (NMR solvents purchased from Cambridge Isotopes Laboratory). Chemical shifts were reported in parts per million (ppm) on the δ scale. Fourier transform infrared (FTIR) spectra of samples as KBr pellets were recorded on a PerkinElmer FTIR 2000 spectrometer in the region of 4000–400 cm−1; 16 scans were signal-averaged with a resolution of 2 cm−1 at room temperature. Gel permeation chromatography (GPC) analyses were performed on a Waters GPC machine at 40 °C, equipped with a 515 HPLC pump, Waters Styragel columns and Waters 2414 refractive index detector. HPLC grade THF was used as the eluent at a flow rate of 1.0 mL min−1. Monodisperse polystyrene standards were used to generate the calibration curve.Hydrogel rheological characterisation
Rheological characterization of the thermogels were carried out as previously described.56–60 PUs synthesised using each catalyst were then formulated into thermogels by mixing the polymers at known weight-to-volume ratios (wt/v%) in deionised water, and agitated using a vortex mixer. The polymer suspensions were left at 2 °C overnight, where they dissolved to form clear transparent solutions. The rheological properties of these solutions and their hydrogels were investigated with a TA Instruments Discovery Hybrid Rheometer Series (DHR-3, Research Instruments, Inc.) with a 40 mm parallel plate geometry, equipped with a temperature-controlled Peltier base plate system. For all measurements, a fixed gap of 900 μm was used. Temperature ramps were conducted from 4 to 40 °C to observe the change in rheological properties of the polyurethane solutions with temperature, at a ramp rate of 5.0 °C min−1, under constant strain of 1.0%, and angular frequency of 10.0 rad s−1. The crossover temperature (Tc) of the solutions was determined as the temperature at which intersection of the storage modulus (G′) and loss modulus (G′′) curves occurred.2D small-angle X-ray scattering (SAXS) experiments
Small-angle X-ray scattering (SAXS) measurements were performed on a Xenocs Xeuss 2.0 instrument at the Institute of Chemical and Engineering Sciences (ICES), A*STAR. 2 wt/v% aqueous solutions of PUs synthesised in the presence of ZDTC, DBTL or in the absence of catalysts were loaded into Hampton 1.5 mm glass capillary tubes which were then sealed with wax. Samples were first cooled to 4 °C at a cooling rate of 10 °C min−1 and held for 10 min to equilibrate. Thereafter, data was collected for 45 min by irradiating the samples with CuKα radiation at a sample-to-detector distance of 1210 mm, which provided a q-range of 0.009–0.21 Å−1. The samples were then warmed at a heating rate of 1 °C min−1 to 12, 25 and 37 °C, and held for 10 min for thermal equilibration before data collection. The 2D SAXS data was reduced and background corrected using Xenoc's data processing and reduction program, Foxtrot (Synchrotron Soleil). The data were fitted in Igor Pro 6.37 using the fitting procedures provided by NIST.61,62 Scattering patterns were adequately described by model fits to the Debye–Anderson–Brumberger (DAB) scattering function,63 which describes a randomly-distributed two-phase system comprised of the dissolved polymer and the aqueous solution or spherical micelles with a Schulz distribution in polydispersity.64Cell viability assay
NIH3T3 cells were cultured in DMEM media supplemented with 10% FBS and 1% penicillin–streptomycin. For cell seeding, the supernatant of a confluent flask (25 cm2) of NIH3T3 was first removed and the cell layer is rinsed twice with 3 mL of sterile 1× phosphate buffered saline (PBS) and the washing solution aspirated. 1 mL of 0.05% trypsin–EDTA was added to the confluent cell layer and incubated for 7 min at 37 °C in the incubator to detach the cell layer. 4 mL of fresh DMEM was added into the culture flask to inactivate the trypsin enzyme and the cell solution is transferred to a 15 mL falcon tube. The cells are pelleted down by centrifuging at 1200 rpm for 5 min and the supernatant decanted. The cell pellet was resuspended in 10 mL of fresh DMEM. 10 μL of the resuspended cell solution was sampled out and the concentration of cells was calculated via a haemocytometer. The concentration of cells in solution is then adjusted to 105 cells per mL and plated into 96 well-plate with 100 μL per well. The seeded cells were then incubated at 37 °C under a 95% air and 5% CO2 atmosphere for 24 h. The cell media was removed and replaced with 100 μL fresh DMEM after 24 h of incubation. Poly(PEG/PPG/PCL urethane) (EPC) samples were prepared by dissolving the polymer in sterile 1× PBS at 2 wt/v%, diluting down to the 10.0, 2.0 and 0.6 mg m−1. Samples were sterilized with UV-irradiation (3 min). 100 μL of the sample solution was added to the DMEM solution in the 96 well plate, and incubated for a further 24 h. 20 μL of Cell titer blue reagent is added to each well and incubated for 1.5 h, and fluorescence of the cell titer blue reagent per well is recorded at 560 nm excitation and 590 nm emission using an InfiniteM200 (Tecan) plate reader. The polymer samples had 8 replicates per concentration, while the control column was free of polymer and contained 1× sterile PBS. The relative cell viability after incubation with the various EPC solutions was obtained by comparing their fluorescence intensity to that of the control column. The cell viability data is expressed as mean ± standard error of the mean (SEM). Cell viability results were further analysed to establish if differences in cell viabilities were significant by employing one-way ANOVA test with Tukey's multiple comparison post hoc test and a level of p < 0.05 was accepted as statistically significant.Results and discussion
Polyurethane synthesis catalysed by ZDTC
With the goal of synthesizing PU thermogelling polymers suitable for biomedical applications,65–75 a model system comprising of PEG2050-diol and PPG2000-diol macromonomers (2:1 wt/wt ratio) was reacted with hexamethylene diisocyanate (HMDI) (OH:NCO mole ratio of 1:1) in toluene at 110 °C for 24 h in the presence of different catalysts (Scheme 1).‡ The PU formed using ZDTC as a catalyst was compared with those using other soluble zinc(II)-compounds under identical reaction conditions, namely zinc(II) neodecanoate, bearing long aliphatic hydrocarbon chains, and zinc(II) acetylacetonate. Organometallic catalysts DBTL and bismuth(III) neodecanoate,39 as well as the organocatalyst DBU, which were shown to be highly effective in catalysing PU synthesis,24 were used as references. To assess the thermogelling property of each amphiphilic PU synthesised by individual catalysts, the resulting polymers were then dissolved in water to form solutions at 10 wt/v%. The temperature at which the polymer solution phase changes from a liquid solution to a solid-like gel, known as the crossover temperature (Tc), was then determined by rheological measurements, together with the storage moduli (G′) and complex viscosities (η*) of the hydrogels at physiological temperature (37 °C). The properties of the polyurethanes and resulting thermogels are summarized in Table 1.
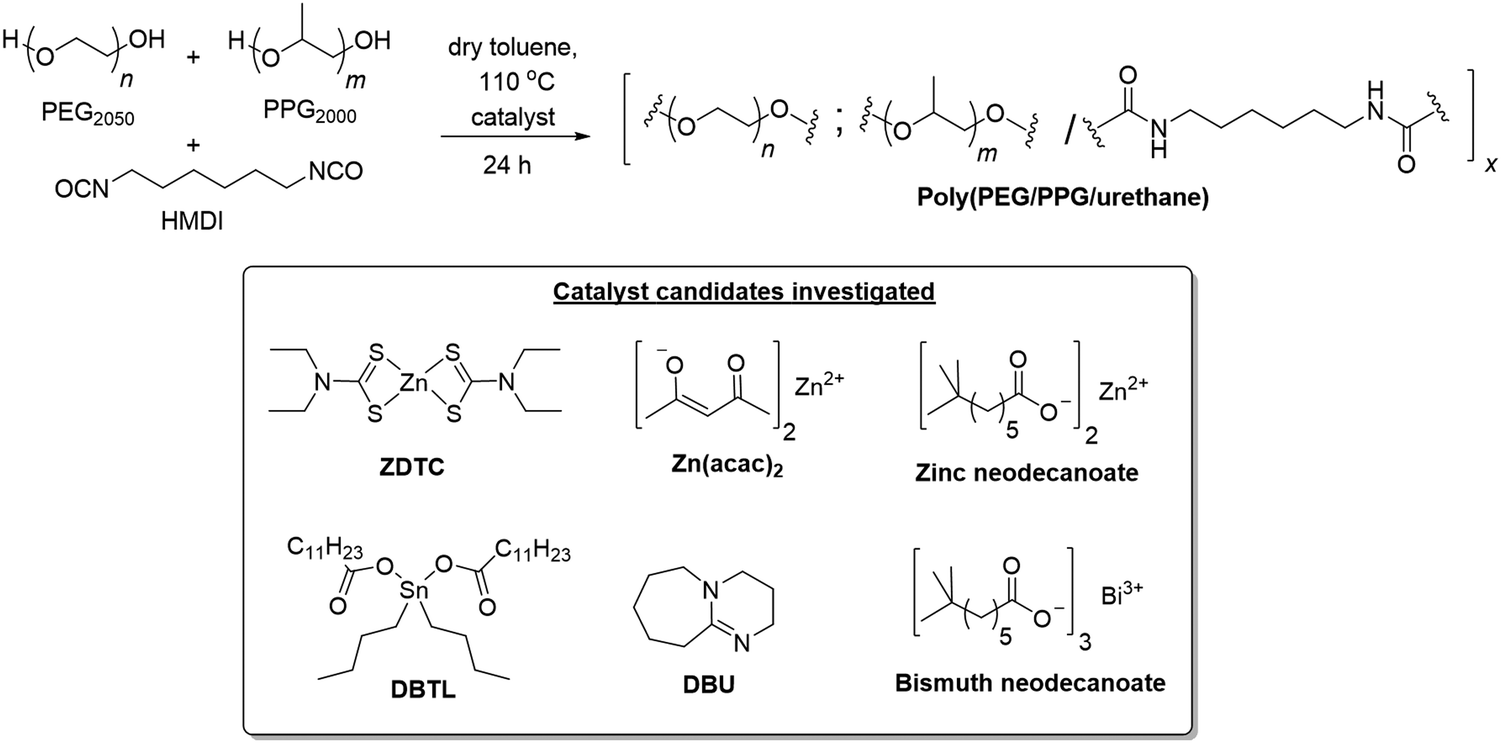 | ||
| Scheme 1 Synthesis of thermogelling polyurethanes and structures of the various catalyst candidates investigated herein. | ||
| S/N | Catalyst | Polymer yieldb/% | Polymer properties | Hydrogel (10 wt/v% PU) rheological propertiese | ||||
|---|---|---|---|---|---|---|---|---|
| PEG:HMDI:PPG molar ratioc |
M n /kg mol−1 | Đ | T c/°C | G′ (37 °C)/Pa | η* (37 °C)/Pa s | |||
|
a All reactions stirred for 24 h at 110 °C in anhydrous toluene under an Ar atmosphere, catalyst loading at 0.67 mol% with respect to each reactive OH/NCO pair.
b Yields of the purified PUs obtained after dialysis for 3 days (MWCO 3500) and freeze-drying.
c Determined from 1H NMR spectroscopy in CDCl3 by integrating the multiplets at 1.08–1.14 ppm for PPG, 3.10–3.15 ppm for HMDI and 1.08–1.14 ppm for PEG. Ideal = 2:3:1.
d Determined from GPC analysis in THF against polystyrene standards (see Fig. S2, ESI).
e Hydrogels containing 10 wt/v% of PU in water, determined by rheological experiments (see Fig. S3, ESI) for oscillatory temperature sweeps.
|
||||||||
| 1 | ZDTC | 91 | 1.9:3.2:1.0 |
62.6 | 1.49 | 8.7 | 4951.2 | 497.0 |
| 2 | Zn(neodec)2 | 61 | 2.4:4.0:1.0 |
29.3 | 1.54 | 37.1 | 64.9 | 9.2 |
| 3 | Zn(acac)2 | 71 | 2.3:3.8:1.0 |
31.8 | 1.52 | 29.4 | 145.2 | 17.2 |
| 4 | DBTL | 83 | 2.1:3.0:1.0 |
34.9 | 1.43 | 21.9 | 558.8 | 58.9 |
| 5 | DBU | 52 | 2.5:3.4:1.0 |
29.2 | 1.39 | 27.3 | 217.9 | 24.9 |
| 6 | Bi(neodec)3 | 66 | 2.7:4.0:1.0 |
30.7 | 1.46 | 38.2 | 67.8 | 9.8 |
| 7 | — | 24 | 6.5:1.0:4.7 |
19.4 | 1.36 | >40 | — | — |
In all cases, as the polyaddition reactions between the diol macromonomers and diisocyanates proceeded, an accompanying increase in solution viscosity was observed with time. Amongst the three zinc(II) catalyst candidates, only the PU synthesis catalysed by ZDTC was found to form a highly-viscous solution after 24 h, whilst only slight increases in solution viscosity were seen in the reactions containing Zn(acac)2 and Zn(neodecanoate)2. While the reference reaction catalyzed by DBTL showed considerable viscosity, it was less viscous than that containing ZDTC. GPC analyses revealed that ZDTC formed polymers with the highest molecular weight (Mnca. 63 kDa, Table 1, entry 1), whereas both Zn(neodecanoate)2 and Zn(acac)2 formed polymers of only approximately 30 kDa (Table 1, entries 2 and 3), corroborating the observed viscosities of the respective reaction mixtures. Even DBTL, DBU and Bi(neodec)3 yielded PUs (Mnca. 35 kDa, Table 1, entries 4–6) considerably shorter than those by ZDTC under identical reaction conditions. In the absence of any added catalyst (Table 1, entry 7), the PUs formed were far inferior to any of the catalyst candidates, with Mn < 20 kDa.
1H NMR spectroscopic analysis of the ZDTC-catalysed PUs show complete incorporation of the macromonomers into the polymer structure, with a PEG:HMDI:PPG molar ratio of close to the ideal 2:3:1. Notably, the PU show identical 1H NMR spectra as the polymer catalyzed by DBTL (Fig. 1A), and FT-IR spectroscopy showed the unequivocal presence of urethane linkages at (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O = 1740 and 1690 cm−1) and absence of the isocyanate functionality (νNCO = 2250 cm−1) (Fig. 1B),20 corroborated by the absence of diagnostic isocyanate 13C NMR resonances of between 120–130 ppm (Fig. S1A, ESI†). 13C NMR spectroscopy also clearly showed that urethane formation is the dominant reaction, evidenced by its prominent resonances at 156 ppm. Like the PUs catalysed by DBTL (Fig. S1B, ESI†), 13C NMR resonances arising from the urea functionality, which form from isocyanates under wet conditions, as well as those of allophanates,76 are notably absent. With the exception of PUs synthesised using ZDTC and DBTL, those from other catalysts showed a slight excess of PEG compared to PPG incorporated into their polymer structure, which may be a consequence of the primary alcohol of PEG-diol reacting at a faster rate than the secondary alcohol of PPG-diol. Unsurprisingly, the largest deviations from the ideal PEG:HMDI:PPG molar ratio was seen for the PUs from the uncatalyzed reaction, indicating only partial reaction completion.
O = 1740 and 1690 cm−1) and absence of the isocyanate functionality (νNCO = 2250 cm−1) (Fig. 1B),20 corroborated by the absence of diagnostic isocyanate 13C NMR resonances of between 120–130 ppm (Fig. S1A, ESI†). 13C NMR spectroscopy also clearly showed that urethane formation is the dominant reaction, evidenced by its prominent resonances at 156 ppm. Like the PUs catalysed by DBTL (Fig. S1B, ESI†), 13C NMR resonances arising from the urea functionality, which form from isocyanates under wet conditions, as well as those of allophanates,76 are notably absent. With the exception of PUs synthesised using ZDTC and DBTL, those from other catalysts showed a slight excess of PEG compared to PPG incorporated into their polymer structure, which may be a consequence of the primary alcohol of PEG-diol reacting at a faster rate than the secondary alcohol of PPG-diol. Unsurprisingly, the largest deviations from the ideal PEG:HMDI:PPG molar ratio was seen for the PUs from the uncatalyzed reaction, indicating only partial reaction completion.
The PUs synthesised using each catalyst were then dissolved in deionised water at 2 °C overnight to form transparent solutions. Upon warming to 20 °C, only the ZDTC-catalysed PUs formed strong transparent gels (Table 1, entry 1), whilst the PUs synthesised using all other catalysts, including DBTL, remained as sols with varying degrees of viscosity. Subsequently, rheological studies were performed on these 10 wt/v% gels by oscillatory temperature sweeps to determine the thermo-responsiveness of the polymer solutions. As shown in the rheological profile of the ZDTC-catalysed PU gel as a function of temperature in Fig. 2, the loss modulus (G′′) of the solution is larger than its storage modulus (G′) at low temperatures as the solution remains in a liquid state. As temperature rises, both G′ and G′′ increase, with the former increasing at a faster rate than the latter as the viscosity of the solution increases. When the solution transits from a liquid state to a semi-solid gel state at Tc, the magnitude of G′ becomes larger than that of G′′ and represents the onset of gelation. Generally, PUs with greater Mn were able to form gels with lower Tc, with higher G′ and viscosities at 37 °C (Table 1). Amongst all the zinc(II) catalyst candidates screened, only the ZDTC-PUs showed a gelling temperature significantly lower than DBTL-PUs, with the former gels at 37 °C showing superior mechanical properties than the latter. In contrast, the aqueous solutions of the PUs synthesised in the absence of catalysts remained as a sol even at 40 °C. Varying the polymer content in hydrogel formulations revealed that at concentrations as low as 1.5 wt/v%, the ZDTC-PUs were still able to gelate at ambient temperature, albeit forming a weak gel, whilst DBTL-PUs only gelate below physiological temperature (37 °C) at concentrations above 5.0 wt/v%. Impressively, as shown in Fig. 3, the ZDTC-PU gel at 1.5 wt% has a considerably lower gelation temperature than the DBTL-catalysed PU at 10.0 wt/v%, with comparable higher storage modulus (G′) at 37 °C (Table S1, ESI†).
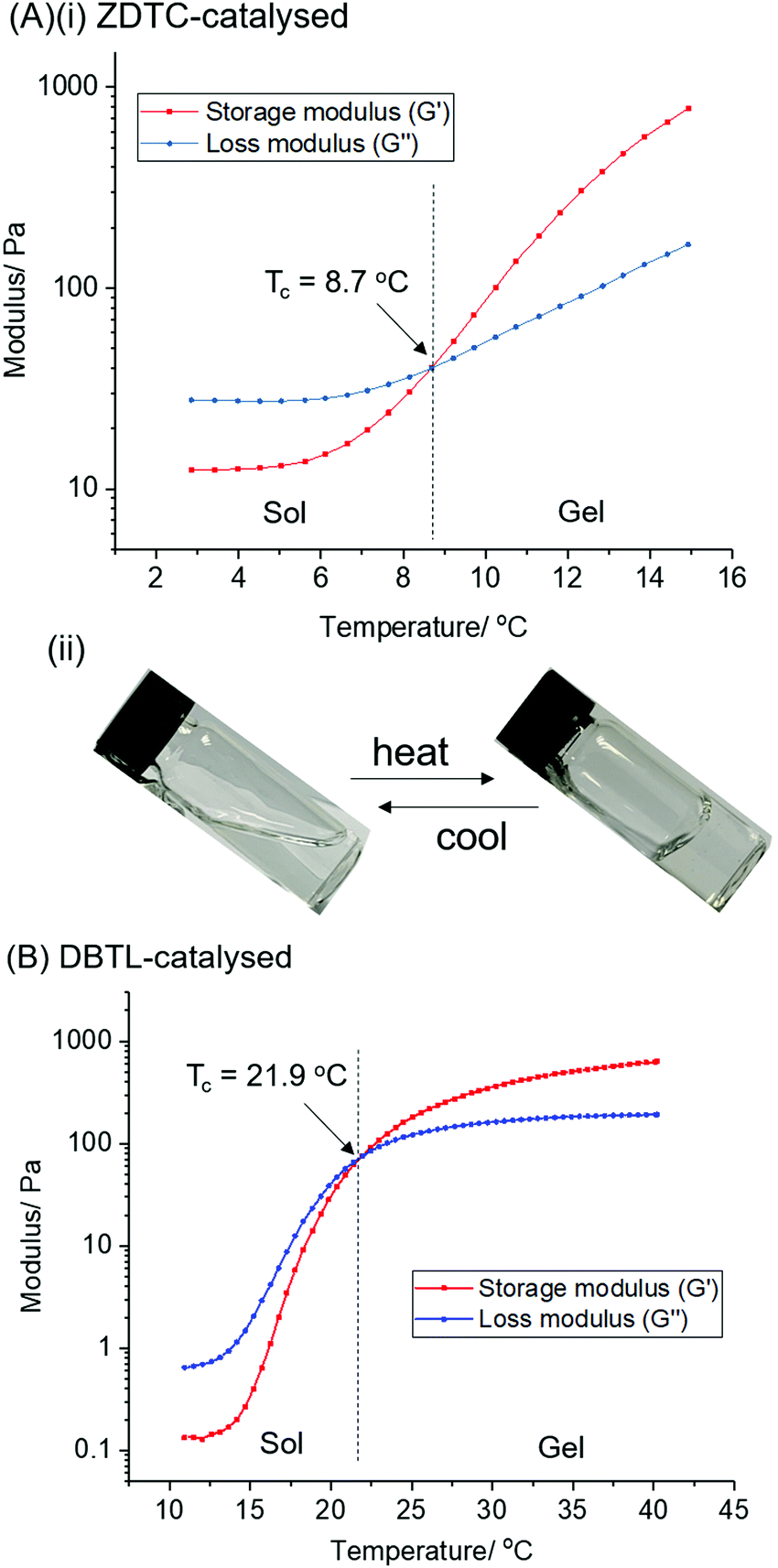 | ||
| Fig. 2 Oscillatory temperature sweep showing the changes in storage (G′) and loss modulus (G′′) as a function of temperature, as well as the gelation temperature of 10 wt% PU catalysed by (A) ZDTC and (B) DBTL. Figure (A)(ii) shows the reversible sol–gel transition of a transparent 10 wt/v% solution of ZDTC-catalysed PU in water. | ||
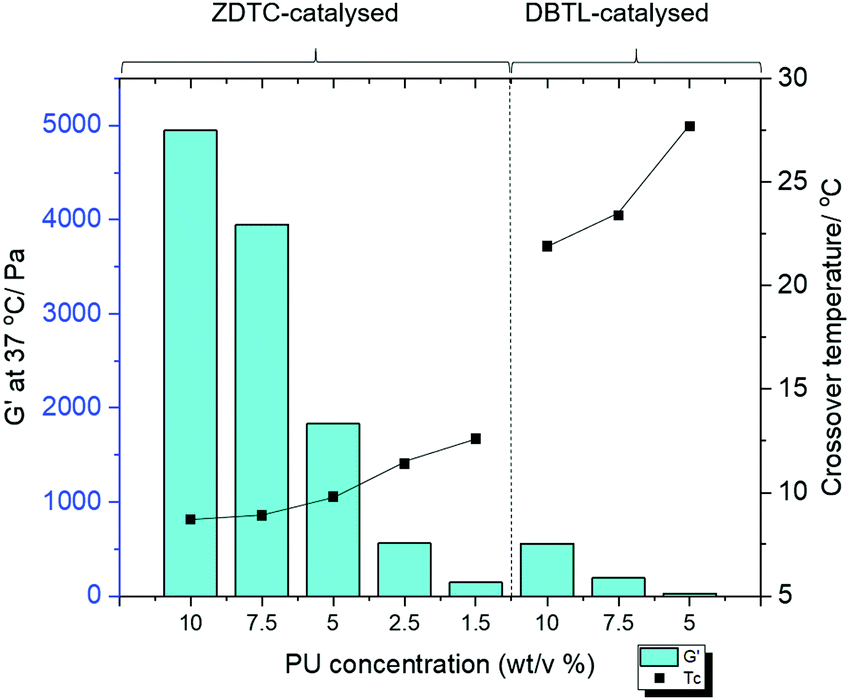 | ||
| Fig. 3 Chart showing the storage moduli (G′, left vertical axis, bars) and crossover temperatures (Tc, right vertical axis, filled dots) of different concentrations of PU, whose syntheses are catalysed by either ZDTC or DBTL. At concentrations of <5.0 wt/v%, the DBTL-catalysed PUs do not form gels below 40 °C. | ||
Having established the potency of ZDTC for PU synthesis, we varied the reaction temperature and solvent to study their influences on the catalysis (Table S2, ESI†). Reducing the reaction temperature from 110 °C to 90 °C resulted in slightly reduced PU molecular weight, correspondingly resulting in slightly weaker gels. However, a further reduction in reaction temperature to 70 °C resulted in a dramatic reduction in PU molecular weight, such that a solution of the polymers at 2.5 wt/v% were unable to gelate at 40 °C. This suggested that the ZDTC catalyst could only be activated when heated above 70 °C. When the reaction was performed at 110 °C using DMF, xylenes and chlorobenzene§ as solvent instead of toluene, PUs with high molecular weights (>50 kDa) were obtained in all cases. Correspondingly, the PUs formed hydrogels of comparable excellent mechanical properties at low polymer concentrations of 2.5 wt%. DMF in particular, resulted in a highly viscous reaction mixture within the first two hours, unlike all other solvents studied. Hence, ZDTC-catalysed PU synthesis is tolerant to solvents with different charge-screening (dielectric constant) and Lewis basicities (quantified by their solvent dielectric constant ε and Gutmann donor numbers respectively, Table S2, ESI†).
We performed a series of NMR experiments to shed light on the possible mechanism of action of the ZDTC catalyst. In d7-toluene, the presence of ZDTC gave a small downfield shift of HMDI methylene (–CH2–) proton signals immediately adjacent to the isocyanate group (Fig. S7, ESI†). This is consistent with possible coordination of the Lewis acidic Zn2+ to the isocyanate group, ostensibly via the oxygen atom,77 which enhances the isocyanate's electron-deficiency and causes a downfield perturbation of the adjacent methylene protons. Just like the mechanism accounting for the activity of organotin(IV) catalysts proposed by Bloodworth and Davis,78 the Zn2+-isocyanate interaction may render the isocyanate's carbon atom more electrophilic for attack by the alcohol unit (Scheme 2). The diethyldithiocarbamate ligands (DTC−), on the other hand, may participate in the catalysis by hydrogen bonding with the alcohol units, which increases the electron density and nucleophilicity of the oxygen atoms. The conversion of DTC− to other catalytically-active species at elevated temperatures may be ruled out, as the 1H NMR spectra of ZDTC in d7-toluene before and after heating for 24 h at 110 °C show negligible differences (Fig. S8, ESI†). It is noteworthy also that amongst the three zinc(II) catalyst candidates, ZDTC showed the best catalytic activity. This may be due to a combination of electronic, solubility and steric factors, amongst which ‘hard–soft’ Zn2+–DTC− interactions may result in greater ligand lability than the ‘hard–hard’ coordination between Zn2+ and the oxygen-containing neodecanoate and acetylacetonate ligands. Finally, we postulate that the unexpected observation of ZDTC catalysing formation of higher molecular weight PUs than DBTL may be due to the high moisture sensitivity of the latter catalyst. DBTL's hydrolytic susceptibility40 in the presence of residual moisture in the reaction from both the solvent and diol reactants may be sufficient to convert it to less catalytically-active species. Alternatively, DBTL has been shown to catalyse very rapid reactions between isocyanates and water,79 which effectively causes the NCO:OH ratio to deviate from unity, concomitantly lowering the overall degree of polyaddition. Indeed, polymerisation reactions repeated under non-anhydrous conditions yielded DBTL-catalysed PUs having considerably lower molecular compared to those catalysed by ZDTC under identical conditions (Fig. S9, ESI†). Although it may be hypothesised accordingly that the ZDTC reaction is more moisture-tolerant, a more thorough investigation of ZDTC's catalytic mechanism is warranted and is underway.
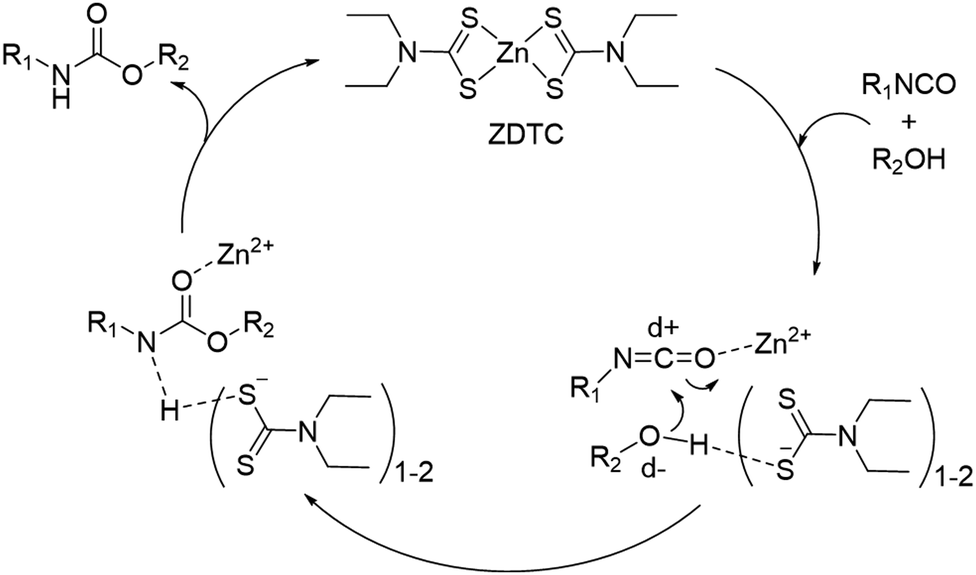 | ||
| Scheme 2 Proposed mechanism of urethane bond formation catalysed by ZDTC. | ||
PEG–PPG block distribution in ZDTC-catalysed polyurethanes
The distribution of hydrophilic (PEG) and hydrophobic (PPG) blocks making up the structure of the polymeric amphiphile exerts profound influences on their self-assembly in water, driven by the hydrophobic effect.4 Hence, we investigated and compared the PEG–PPG block distribution in the PUs catalysed by ZDTC and DBTL via the time-dependent block incorporation within the growing polymer chain. Generally, secondary alcohols (such as those found on PPG) are known to react more slowly with isocyanates compared to primary alcohols (e.g. those in PEG) both in the absence80 and presence81,82 of Lewis acid catalysts. If PEG is preferentially reacted before PPG, this can result in the formation of a tapered diblock-copolymer structure, with a greater number of hydrophilic PEG blocks at one end of the polymer and more hydrophobic PPG blocks at the other. The reactions, containing 2:1:3 molar ratio of PEG, PPG and HMDI were performed at 110 °C in toluene the presence of either catalyst, and at regular reaction durations, a small aliquot of the reaction mixture was extracted and precipitated in hexane, and the oligomer/polymer was isolated by filtration. Due to the miscibility of PPG macromonomer and HMDI reactant with hexane, any unreacted PPG and HMDI were removed from the mixture during filtration, whilst the PPG and HMDI present in the isolated polymer would have been already integrated covalently into the growing polymer chains. As each diol (either PEG or PPG) will react with a HMDI reactant in a 1:1 molar ratio, the temporal evolution of the relative HMDI:PPG ratio in each aliquot can indicate the block structure of the PU. These molar ratios were determined by integrating the appropriate 1H NMR proton resonances for each isolated polymer aliquot. For instance, if PEG reacts much faster than PPG, there will be initially very low PPG incorporation compared to HMDI in the early stages of the reaction, which will gradually increase with reaction duration to reach a theoretical maximum of 0.33 when complete monomer incorporation has been achieved. Conversely, the formation of a completely random block copolymer, without any preference for either PEG or PPG, will have a consistent theoretical PPG-to-HMDI molar ratio of 0.33 regardless of the extent and duration of the reaction.
The evolution of the PPG: HMDI ratios of PUs catalysed by ZDTC as a function of time is shown in Fig. 4, as well as Table S3, ESI.† A distinct increase in PPG: HMDI molar ratio throughout the first hour of reaction, rising from ca. 0.2 after 10 min to approximately 0.3 after an hour, which accompanied an increase in PU molecular weight. This indicated that ZDTC facilitated the preferential reaction between the primary alcohols of PEG and HMDI over the secondary alcohols of PPG, in a similar way as that by DBTL.83 It is hence likely that the ZDTC-catalysed PUs possess non-random PEG–PPG block distribution along the polymer backbone, with certain sites containing a higher proportion of PPG blocks being more hydrophobic than the analogous PEG-rich sites.
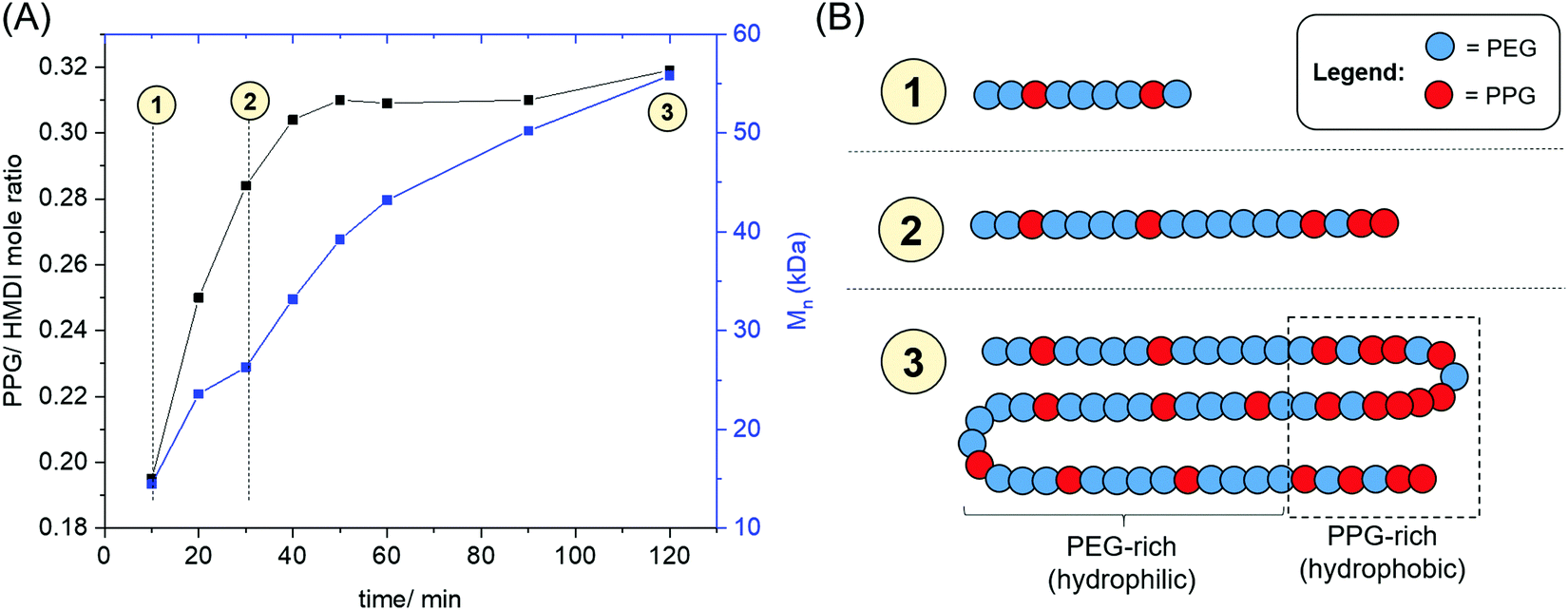 | ||
| Fig. 4 (A) Evolution of the PPG:HMDI molar ratios and GPC molecular weight of ZDTC-catalysed PU as a function of time; (B) illustration of PEG–PPG distribution in the ZDTC-catalysed polymer at different timepoints during the reaction. | ||
Temperature-triggered hierarchical self-assembly of ZDTC-catalysed PU amphiphiles
Amphiphilic block-copolymers can form self-assembled micelles when dissolved in water, driven by the hydrophobic effect. Upon warming, these micelles can further aggregate by hierarchical self-assembly to form hydrogels.4,84 The self-assembly behaviour of polymers are determined by their structures and molecular weights.85 Thus, to shed light on how these differences can affect the thermo-responsive self-assembly of ZDTC- and DBTL-catalysed PUs, which ultimately resulted in their dramatic differences in hydrogel properties shown in Table 1, we characterized their aqueous microstructure using small-angle X-ray scattering (SAXS). Evidence of the polymers’ self-assembly into micelles in water was provided by dynamic light scattering (DLS) experiments (Fig. S10, ESI†) with dilute (0.1 wt/v%) PU solutions at 25 °C, which showed that the ZDTC-PUs formed micelles with a broader size distribution than the DBTL-PUs. The critical micelle concentrations (CMCs) of these PUs were also determined by the dye solubilization method with diphenylhexatriene (DPH) dye at 25 °C. As shown in Fig. 5, the CMC of ZDTC-PUs (0.041 wt/v%) was considerably lower than that of the DBTL-PUs (0.127 wt/v%). This is consistent with the longer ZDTC-PUs, with more well-defined hydrophobic PPG-rich segments increasing the overall hydrophobicity of the polymer, facilitating their self-assembly into micelles at lower concentrations.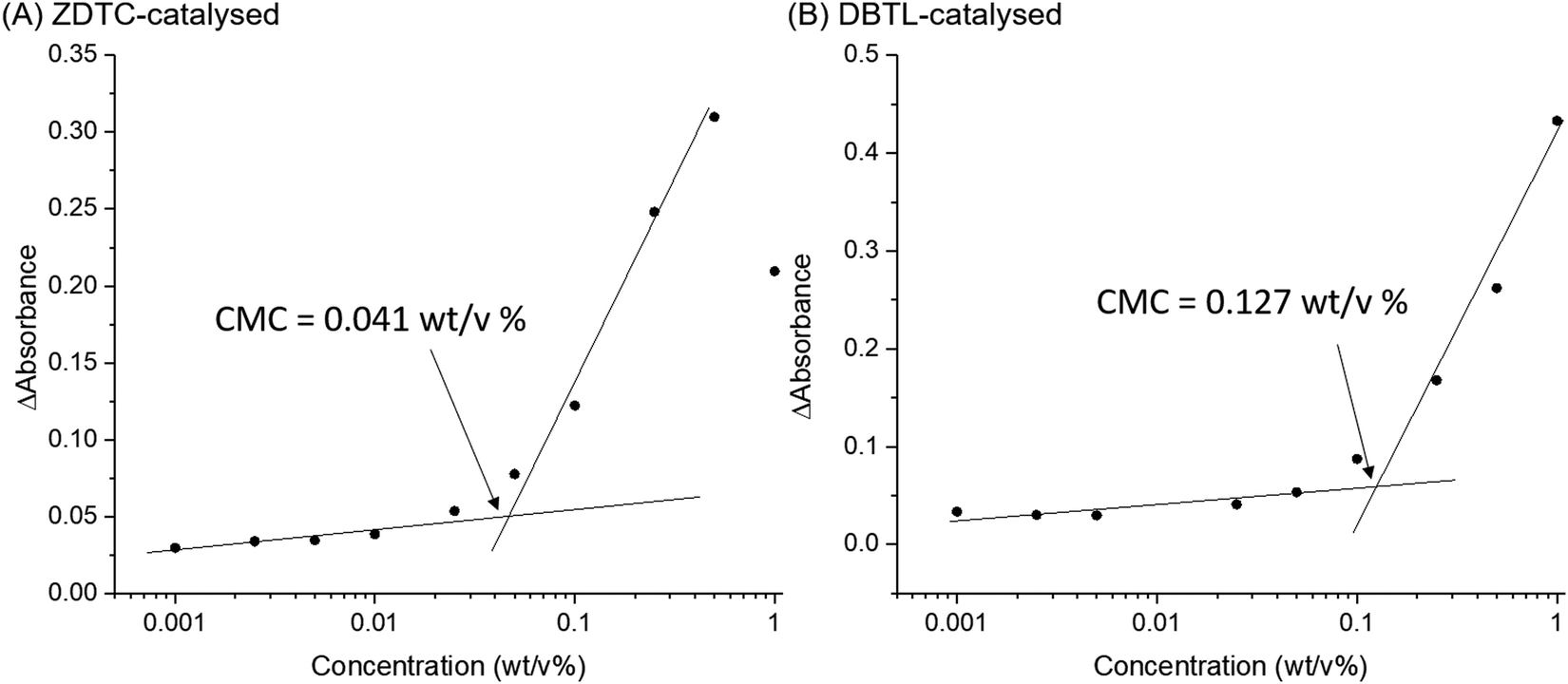 | ||
| Fig. 5 Difference in DPH absorbance at 376 and 394 nm as a function of polymer concentration for PUs catalysed by (A) ZDTC and (B) DBTL. The CMC of the polymer was determined by the intersection of the extrapolated linear best fit lines at low and high concentrations of polymer. | ||
SAXS experiments were performed using 2 wt/v% solutions of PUs catalysed by ZDTC and DBTL at 4, 12, 25 and 37 °C to study the temperature-induced changes in self-assembled microstructures. As a reference, PUs synthesised in the absence of catalyst (Table 1, entry 7) were also probed under identical conditions. As shown in Fig. 6A and B, at low temperatures (4 °C), DBTL-catalysed and reference PU solutions show similar scattering patterns while even at 4 °C, the scattering function for the ZDTC-catalysed PU solution showed a prominent difference in scattering at q < 0.04 Å−1 (Fig. 6C), indicative of some degree of structure and polymer self-assembly even at low temperatures. As the temperature increased, the scattering patterns for the DBTL-PU and reference PU solutions show a change in the shape of the scattering pattern due to changes in microstructure upon gelation. Similar temperature-driven changes in scattering functions for related amphiphilic Pluronic block co-polymers of PEG and PPG have been observed, indicative of micelle formation and their self-assembly by micellar ordering.86–89 For the ZDTC-catalysed PU solution, smaller but significant changes (vide infra) in the scattering pattern was observed at higher temperatures (Fig. 6C).
 | ||
| Fig. 6 Small-angle X-ray scattering patterns of 2 wt/v% aqueous solutions of (A) uncatalyzed PUs (□); (B) DBTL-catalysed PUs (Δ) and (C) ZDTC-catalysed PUs (○) at 4, 12, 25 and 37 °C. The solid black lines are model fits to the data. Curves are offset for clarity. | ||
At 4 °C, the scattering pattern for all three polymers could be modelled using the Debye–Anderson–Brumberger (DAB) scattering function,63 characteristic of a randomly-distributed two-phase system comprised of the dissolved polymer and the aqueous solution. The correlation lengths, which indicates the average spacing between regions of both phases, obtained from the least-squares fit of the experimental data, are summarized in Table 2. Both uncatalysed and DBTL-catalysed PUs are likely to form random networks from entangled unassociated unimers90 at 4 °C. Warming the DBTL-PU solution from 4 °C to 12 °C results in an increase in correlation length, suggestive of some degree polymer self-assembly which increases the average separation between the polymer and water phases. On the other hand, the uncatalysed PU does not show a significant increase in correlation length until 37 °C, which corroborates with its poorer temperature-responsive behavior (see Table 1). Even at 4 °C, the ZDTC-catalysed PUs show a much larger correlation length compared to the other two polymers (Table 2), suggesting a significant degree of polymer self-assembly to form randomly-distributed polymer domains that phase separate from the surrounding aqueous medium.
| 4 °C | 12 °C | 25 °C | 37 °C | ||
|---|---|---|---|---|---|
| a Synthesised in the absence of catalysts (Table 1, entry 8). b Fit to the Debye–Anderson–Brumberger (DAB) scattering model. c Fit to the Schulz spheres model. d No reasonable fit obtained. | |||||
| DBTL-PUs | Correlation lengthb/Å | 21.7 ± 0.4 | 48.3 ± 0.6 | —d | —d |
| Mean radiusc/Å | —d | —d | 95.4 ± 0.9 | 92.0 ± 0.7 | |
| Polydispersityc | —d | —d | 0.29 ± 0.01 | 0.27 ± 0.01 | |
| ZDTC-PUs | Correlation lengthb/Å | 93.7 ± 0.7 | —d | —d | —d |
| Mean radiusc/Å | —d | 89.5 ± 1.0 | 93.4 ± 0.9 | 57.8 ± 1.8 | |
| Polydispersityc | —d | 0.35 ± 0.01 | 0.31 ± 0.01 | 0.57 ± 0.2 | |
| Reference PUsa | Correlation lengthb/Å | 25.6 ± 0.5 | 24.3 ± 0.5 | 29.1 ± 0.5 | 49.5 ± 0.5 |
At 12 °C, following desolvation of the hydrophobic PPG segments in water, further hierarchical self-assembly of the ZDTC-PUs occur, such that its scattering pattern could not be satisfactorily described using the DAB model. Unlike the structurally well-defined triblock Pluronic polymers, which are known to form core–shell micelles with a hydrophobic PPG core and hydrophilic PEG corona,91 the considerably more random PEG–PPG block distribution of the ZDTC- and DBTL-catalysed PUs (vide supra) can be expected to form non-uniform polydisperse micelles containing a distribution of micelle sizes. Satisfactory fits of the scattering functions for ZDTC-PUs at 12, 25 and 37 °C, as well as DBTL-PUs at 25 and 37 °C, were obtained using a model describing polydisperse spheres with a Schulz distribution of radii.64 The mean radius and polydispersity of the micelles are summarized in Table 2.
As seen from Table 2, the resulting polydisperse spherical micelles formed in both ZDTC- and DBTL-catalysed PUs have similar mean radii and polydispersity at 25 °C. Considering the greater molecular weight of the ZDTC-PUs, this result thus suggests that these polymers are able to pack more tightly into the micelles compared to DBTL-PUs. Notably, these micelles are comparable in size to those of Pluronics at similar temperatures,87,88 despite the considerably larger molecular weights of our PUs. This suggests that the individual PU chains may fold back on themselves during their self-assembly to form micelles, such that the randomly-positioned hydrophobic PPG segments along the polymer chain can associate together to form the micellar core. Upon further warming to 37 °C, whilst the mean radii and polydispersity of the DBTL-PU micelles remain roughly unchanged, the ZDTC-PU micelles show significant shrinkage accompanied by an increase in polydispersity (Table 2). We propose that these differences in self-assembly behaviors can be attributed to the longer polymers synthesized using ZDTC compared with DBTL, which possess a greater number of hydrophobic PPG-rich domains. Compared to the shorter DBTL polymers, the ZDTC polymers can interact with each other at a greater number of sites by hydrophobic association. This ultimately leads to stronger polymer–polymer interactions, which drives tighter micelle packing that leads to more pronounced micelle shrinkage as further PPG desolvation occurs upon warming. The same reason likely accounts for the lower CMC observed for the ZDTC-catalysed PUs compared with those from DBTL. Although Pluronic micelles show a slight expansion upon warming,87 which contrasts with the ZDTC-PU micelles, it is important to note that many competing factors affect their temperature-responsive behaviour. For instance, additives such as oils in aqueous Pluronic F127 solutions has been shown to result in micelle core shrinkage upon warming.92 For the ZDTC-PUs herein, the presence of urethane linkages which are capable of inter-and intramolecular hydrogen bonding interactions,93–95 can likely affect polymer and micelle hierarchical self-assembly significantly. This contrasts with Pluronics which are solely polyethers and do not possess hydrogen bonding units. Indeed, upon desolvation in the PPG-rich polymer domains, the resulting more hydrophobic environment can augment the strength of intermolecular polymer–polymer hydrogen bonding interactions,96 as competition from water molecules (which are strong hydrogen bond acceptors)97 is greatly reduced, further enhancing polymer packing.
Cell viability studies
The contamination of polymers by metal catalyst residues used in their synthesis can impact their physical properties,98 biosafety and commercial applications.99 However, despite significant research efforts for their removal,99,100 this can be difficult to achieve and the cost is often prohibitive.23 To study the possible cytotoxic effects of residual ZDTC and DBTL catalysts in our thermogelling PUs, we performed cell viability studies on crude, unpurified polymers immediately after synthesis. In this study, we synthesised three-component PUs using PEG2050, PPG2000 and PCL2000 building blocks (see ESI† for details) which have been previously shown to be highly biocompatible in vivo and in vitro,17 using identical loadings of ZDTC and DBTL. The cell viability studies were performed using aqueous PU solutions (10.0, 2.0 and 0.6 mg mL−1) with the cell titer blue assay which measures the metabolic viability of murine NIH 3T3 fibroblast cells. In viable cells, the assay's indicator dye resazurin is reduced to fluorescent resorufin, allowing the fluorescent intensity to be representative of the number of metabolically viable cells.The results of the cell viability study are summarised in Fig. 7, which show that cells incubated with DBTL-catalysed PU exhibited low cell viability at all three polymer concentrations, even as low as 0.6 mg mL−1 (0.06 wt/v%), showing that the remnant tin catalyst present in the polymer after precipitation was still sufficient to induce significant cytotoxicity. In contrast, while ZDTC-catalysed PUs showed some loss of cell viability at a concentration of 10 mg mL−1, it was less toxic than its DBTL counterpart. At lower polymer concentrations of 2 and 0.6 mg mL−1, high cell viability was observed, whose difference from the positive control (without PUs) was statistically insignificant. As seen, the lower cytotoxicity of the ZDTC-catalysed unpurified PUs compared to DBTL is beneficial for biomedical applications, whilst simultaneously reducing the environmental impact from the synthesis process.
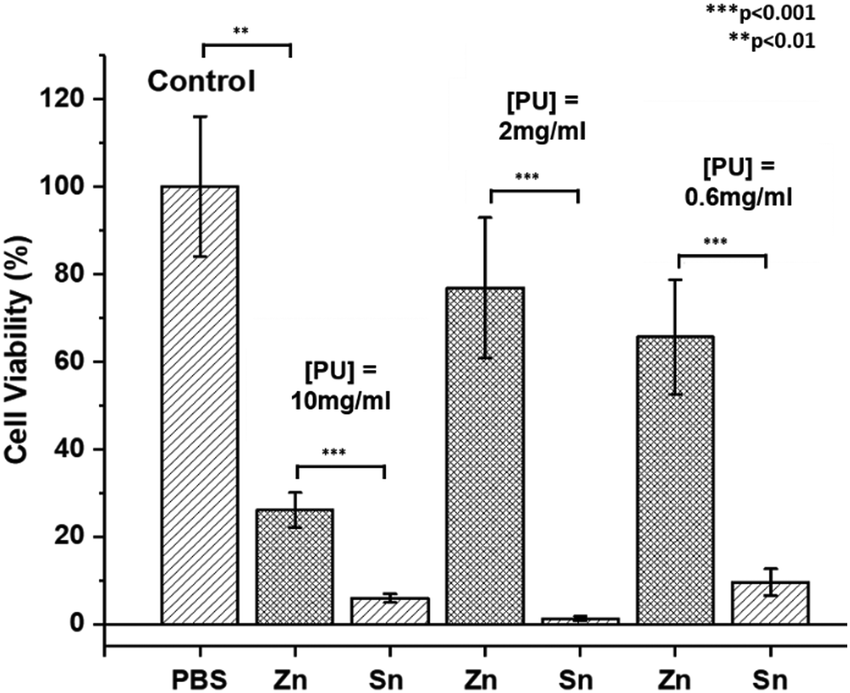 | ||
| Fig. 7 Bar chart comparing the viability of murine NIH 3T3 fibroblast cells incubated with unpurified PU synthesised using either ZDTC (Zn) or DBTL (Sn) catalyst at PU concentrations of 10.0, 2.0 and 0.6 mg mL−1. The positive control experiments were performed in the absence of polymers (PBS) for comparison. Cell viability values are the average of 4 repeats with the error bars indicating the standard error of the mean. | ||
Conclusions
In conclusion, we have demonstrated a promising alternative catalytic application of ZDTC for the synthesis of polyurethanes suitable for use as biocompatible thermogels, in addition to their current well-established use as catalysts for vulcanisation of rubber. Like the commonly-used DBTL catalyst for PUs, ZDTC can synthesise high molecular weight PUs under identical reaction conditions. Uniquely, the surprisingly exceptional catalytic properties of ZDTC compared with other organozinc catalysts studied herein allows the reaction to tolerate a range of organic solvents, including the presence of residual water, successfully forming PUs with high molecular weights in all cases. Our results show that ZDTC catalysis likely favours the formation of non-random block copolymer structures, with the presence of PEG- and PPG-rich hydrophilic and hydrophobic domains. Nonetheless, the ZDTC-catalysed PUs exhibit temperature-responsive sol-to-gel phase transition upon warming, forming mechanically-robust hydrogels at much lower temperatures at considerably reduced polymer concentrations than those synthesised using the DBTL catalyst. SAXS analysis of the microstructure of the PU solutions revealed augmented hierarchical self-assembly of ZDTC-catalysed PUs into micelles at lower temperatures. Valuably, cell viability experiments showed that the unpurified PUs catalysed by ZDTC showed much lower cytotoxicity compared to those made using DBTL. Indeed, the further potential applications of ZDTC as alternatives to other classes of polymerisation reactions mediated by Lewis acidic organotin catalysts, such as polyester synthesis by acid-alcohol polycondensations101 or polysiloxane crosslinking,102 may be explored in the near future.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by an IAF-PP (HMBS Domain) grant H17/01/a0/013 (OrBID): OculaR BIomaterials and Device. The authors would like to acknowledge Chia Sze Chen (ICES) for help with GPC analysis, as well as Yeo Wen Cong (ICES) and Lim Chen Chuan (ICES) for assistance in SAXS data acquisition.References
- H.-W. Engels, H.-G. Pirkl, R. Albers, R. W. Albach, J. Krause, A. Hoffmann, H. Casselmann and J. Dormish, Angew. Chem. Int. Ed., 2013, 52, 9422–9441 CrossRef CAS.
- W. Wang and C. Wang, in The Design and Manufacture of Medical Devices, ed. J. P. Davim, Woodhead Publishing, 2012, pp. 115–151, DOI:10.1533/9781908818188.115.
- J. Joseph, R. M. Patel, A. Wenham and J. R. Smith, Trans. IMF, 2018, 96, 121–129 CrossRef CAS.
- S. S. Liow, Q. Dou, D. Kai, A. A. Karim, K. Zhang, F. Xu and X. J. Loh, ACS Biomater. Sci. Eng., 2016, 2, 295–316 CrossRef CAS.
- L. Polo Fonseca, R. B. Trinca and M. I. Felisberti, Int. J. Pharm., 2018, 546, 106–114 CrossRef CAS.
- J. Y. C. Lim, Q. Lin, K. Xue and X. J. Loh, Mater. Today Adv., 2019, 3, 100021 CrossRef.
- K. Xue, X. Zhao, Z. Zhang, B. Qiu, Q. S. W. Tan, K. H. Ong, Z. Liu, B. H. Parikh, V. A. Barathi, W. Yu, X. Wang, G. Lingam, W. Hunziker, X. Su and X. J. Loh, Biomater. Sci., 2019, 7, 4603–4614 RSC.
- Y. Zhang, J. Ding, D. Sun, H. Sun, X. Zhuang, F. Chang, J. Wang and X. Chen, Mater. Sci. Eng., C, 2015, 49, 262–268 CrossRef CAS.
- S. S. Liow, Q. Dou, D. Kai, Z. Li, S. Sugiarto, C. Y. Y. Yu, R. T. K. Kwok, X. Chen, Y.-L. Wu, S. T. Ong, A. Kizhakeyil, N. K. Verma, B. Z. Tang and X. J. Loh, Small, 2017, 13, 1603404 CrossRef.
- X. J. Loh, S. H. Goh and J. Li, Biomaterials, 2007, 28, 4113–4123 CrossRef CAS.
- B. Jeong, Y. H. Bae, D. S. Lee and S. W. Kim, Nature, 1997, 388, 860–862 CrossRef CAS.
- X. Fan, J. Y. Chung, Y. X. Lim, Z. Li and X. J. Loh, ACS Appl. Mater. Interfaces, 2016, 8, 33351–33370 CrossRef CAS.
- A. A. Karim, Q. Dou, Z. Li and X. J. Loh, Chem. – Asian J., 2016, 11, 1300–1321 CrossRef.
- J. Pushpamalar, A. K. Veeramachineni, C. Owh and X. J. Loh, ChemPlusChem, 2016, 81, 504 CrossRef CAS.
- X. J. Loh, J. Appl. Polym. Sci., 2013, 127, 992–1000 CrossRef CAS.
- X. J. Loh, W. C. D. Cheong, J. Li and Y. Ito, Soft Matter, 2009, 5, 2937–2946 RSC.
- Z. Liu, S. S. Liow, S. L. Lai, A. Alli-Shaik, G. E. Holder, B. H. Parikh, S. Krishnakumar, Z. Li, M. J. Tan, J. Gunaratne, V. A. Barathi, W. Hunziker, R. Lakshminarayanan, C. W. T. Tan, C. K. Chee, P. Zhao, G. Lingam, X. J. Loh and X. Su, Nat. Biomed. Eng., 2019, 3, 598–610 CrossRef CAS.
- K. K. Majumdar, A. Kundu, I. Das and S. Roy, Appl. Organomet. Chem., 2000, 14, 79–85 CrossRef CAS.
- S.-G. Luo, H.-M. Tan, J.-G. Zhang, Y.-J. Wu, F.-K. Pei and X.-H. Meng, J. Appl. Polym. Sci., 1997, 65, 1217–1225 CrossRef CAS.
- E. Delebecq, J.-P. Pascault, B. Boutevin and F. Ganachaud, Chem. Rev., 2013, 113, 80–118 CrossRef CAS.
- B. J. Brüschweiler, F. E. Würgler and K. Fent, Aquat. Toxicol., 1995, 32, 143–160 CrossRef.
- D. J. Coady, H. W. Horn, G. O. Jones, H. Sardon, A. C. Engler, R. M. Waymouth, J. E. Rice, Y. Y. Yang and J. L. Hedrick, ACS Macro Lett., 2013, 2, 306–312 CrossRef CAS.
- H. Sardon, A. Pascual, D. Mecerreyes, D. Taton, H. Cramail and J. L. Hedrick, Macromolecules, 2015, 48, 3153–3165 CrossRef CAS.
- J. Alsarraf, Y. A. Ammar, F. Robert, E. Cloutet, H. Cramail and Y. Landais, Macromolecules, 2012, 45, 2249–2256 CrossRef CAS.
- O. Coutelier, M. El Ezzi, M. Destarac, F. Bonnette, T. Kato, A. Baceiredo, G. Sivasankarapillai, Y. Gnanou and D. Taton, Polym. Chem., 2012, 3, 605–608 RSC.
- D. K. Chattopadhyay and K. V. S. N. Raju, Prog. Polym. Sci., 2007, 32, 352–418 CrossRef CAS.
- H. Sardon, A. C. Engler, J. M. W. Chan, J. M. García, D. J. Coady, A. Pascual, D. Mecerreyes, G. O. Jones, J. E. Rice, H. W. Horn and J. L. Hedrick, J. Am. Chem. Soc., 2013, 135, 16235–16241 CrossRef CAS.
- M. Muuronen, P. Deglmann and Ž. Tomović, J. Org. Chem., 2019, 84, 8202–8209 CrossRef CAS.
- W. J. Blank, Z. A. He and E. T. Hessell, Prog. Org. Coat., 1999, 35, 19–29 CrossRef CAS.
- Y. Schellekens, B. Van Trimpont, P.-J. Goelen, K. Binnemans, M. Smet, M.-A. Persoons and D. De Vos, Green Chem., 2014, 16, 4401–4407 RSC.
- D. B. Hogan, C. Harbidge and A. Duncan, Can. Geriatr. J., 2018, 21, 307–309 CrossRef.
- S. L. O'Neal and W. Zheng, Curr. Environ. Health Rep., 2015, 2, 315–328 CrossRef.
- L. M. Gaetke, H. S. Chow-Johnson and C. K. Chow, Arch. Toxicol., 2014, 88, 1929–1938 CrossRef CAS.
- M. Costa and A. Murphy, in The Nutritional Biochemistry of Chromium (III) (Second Edition), ed. J. B. Vincent, Elsevier, 2019, pp. 341–359, DOI:10.1016/B978-0-444-64121-2.00011-8.
- S. A. Katz and H. Salem, J. Appl. Toxicol., 1993, 13, 217–224 CrossRef CAS.
- P. B. Tchounwou, C. G. Yedjou, A. K. Patlolla and D. J. Sutton, Exp. Suppl., 2012, 101, 133–164 Search PubMed.
- Y.-j. Hong, W. Liao, Z.-f. Yan, Y.-c. Bai, C.-l. Feng, Z.-x. Xu and D.-y. Xu, J. Chem., 2020, 9010348 CAS.
- J. Y. C. Lim, S. S. Goh, S. S. Liow, K. Xue and X. J. Loh, J. Mater. Chem. A, 2019, 7, 18759–18791 RSC.
- R. Cannas and U. Burckhardt, Bismuth containing catalysts for polyurethane compositions, Switzerland Pat., EP2604615A1, 2019 Search PubMed.
- M. S. Rolph, A. L. J. Markowska, C. N. Warriner and R. K. O'Reilly, Polym. Chem., 2016, 7, 7351–7364 RSC.
- S. Enthaler and X.-F. Wu, in Zinc Catalysis: Applications in Organic Synthesis, First Edition, ed. S. Enthaler and X.-F. Wu, Wiley-VCH Verlag GmbH & Co. KGaA, 1st edn, 2015, ch. 1, pp. 1–4 Search PubMed.
- S. Ní Shúilleabháin, C. Mothersill, D. Sheehan, N. M. O’Brien, J. O’Halloran, F. N. A. M. Van Pelt and M. Davoren, Toxicol. In Vitro, 2004, 18, 365–376 Search PubMed.
- S. Frassinetti, G. L. Bronzetti, L. Caltavuturo, M. Cini and C. D. Croce, J. Environ. Pathol. Toxicol. Oncol., 2006, 25, 597–610 CrossRef CAS.
- J. Bai, X. Xiao, Y. Zhang, J. Chao and X. Chen, Dalton Trans., 2017, 46, 9846–9858 RSC.
- R. Duan, C. Hu, Z. Sun, X. Pang and X. Chen, ACS Omega, 2018, 3, 11703–11709 CrossRef CAS.
- M. van Meurs, G. J. P. Britovsek, V. C. Gibson and S. A. Cohen, J. Am. Chem. Soc., 2005, 127, 9913–9923 CrossRef CAS.
- S. S. Park, C. S. Kim, S. D. Kim, S. J. Kwon, H. M. Lee, T. H. Kim, J. Y. Jeon and B. Y. Lee, Macromolecules, 2017, 50, 6606–6616 CrossRef CAS.
- Y. Zhao, L. Wang, H. Yu, G. Jing, C. Li, Y. Chen and M. Saleem, J. Polym. Res., 2014, 21, 470 CrossRef.
- G. Trott, J. A. Garden and C. K. Williams, Chem. Sci., 2019, 10, 4618–4627 RSC.
- A. Thevenon, C. Romain, M. S. Bennington, A. J. P. White, H. J. Davidson, S. Brooker and C. K. Williams, Angew. Chem. Int. Ed., 2016, 55, 8680–8685 CrossRef CAS.
- B. Bantu, G. Manohar Pawar, K. Wurst, U. Decker, A. M. Schmidt and M. R. Buchmeiser, Eur. J. Inorg. Chem., 2009, 1970–1976 CrossRef CAS.
- S. Mostoni, P. Milana, B. Di Credico, M. D’Arienzo and R. Scotti, Catalysts, 2019, 9 Search PubMed.
- M. N. Alam, S. K. Mandal and S. C. Debnath, Rubber Chem. Technol., 2012, 85, 120–131 CrossRef CAS.
- P. J. Nieuwenhuizen, A. W. Ehlers, J. G. Haasnoot, S. R. Janse, J. Reedijk and E. J. Baerends, J. Am. Chem. Soc., 1999, 121, 163–168 CrossRef CAS.
- X. Wu, J. A. Smith, S. Petcher, B. Zhang, D. J. Parker, J. M. Griffin and T. Hasell, Nat. Commun., 2019, 10, 647 CrossRef.
- G. Barouti, S. S. Liow, Q. Dou, H. Ye, C. Orione, S. M. Guillaume and X. J. Loh, Chem. – Eur. J., 2016, 22, 10501–10512 CrossRef CAS.
- X. J. Loh, B. J. H. Yee and F. S. Chia, J. Biomed. Mater. Res., Part A, 2012, 100, 2686–2694 CrossRef.
- X. J. Loh, V. P. N. Nguyen, N. Kuo and J. Li, J. Mater. Chem., 2011, 21, 2246–2254 RSC.
- Y.-L. Wu, H. Wang, Y.-K. Qiu and X. J. Loh, RSC Adv., 2016, 6, 44506–44513 RSC.
- V. P. N. Nguyen, N. Kuo and X. J. Loh, Soft Matter, 2011, 7, 2150–2159 RSC.
- B. Hammouda, in Polymer Characteristics, Springer Berlin Heidelberg, Berlin, Heidelberg, 1993, pp. 87–133, DOI:10.1007/BFb0025862.
- S. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- P. Debye, H. R. Anderson and H. Brumberger, J. Appl. Phys., 1957, 28, 679–683 CrossRef CAS.
- G. V. Schulz, Phys. Chem., 1935, 43, 25 Search PubMed.
- X. J. Loh, L. W. I. Cheng and J. Li, Macromol. Symp., 2010, 296, 161–169 CrossRef CAS.
- X. J. Loh, H. X. Gan, H. Wang, S. J. E. Tan, K. Y. Neoh, S. S. Jean Tan, H. F. Diong, J. J. Kim, W. L. Sharon Lee, X. Fang, O. Cally, S. S. Yap, K. P. Liong and K. H. Chan, J. Appl. Polym. Sci., 2014, 131 Search PubMed.
- L. Jiang, Z. Luo, X. J. Loh, Y.-L. Wu and Z. Li, ACS Appl. Bio Mater., 2019, 2, 3591–3600 CrossRef CAS.
- Z.-X. Zhang, S. S. Liow, K. Xue, X. Zhang, Z. Li and X. J. Loh, ACS Appl. Polym. Mater., 2019, 1, 1769–1777 CrossRef CAS.
- Z. Luo, L. Jiang, S. Yang, Z. Li, W. M. W. Soh, L. Zheng, X. J. Loh and Y. L. Wu, Adv. Healthcare Mater., 2019, 8, 1900406 CrossRef.
- Z. W. K. Low, Y. Luo, K. Zhang, Q. Lin, C. Owh, X. Chen and X. J. Loh, Biomater. Sci., 2020, 8, 960–972 RSC.
- Z. Luo, K. Xue, X. Zhang, J. Y. Lim, X. Lai, D. J. Young, Z.-X. Zhang, Y.-L. Wu and X. J. Loh, Biomater. Sci., 2020, 8, 1364–1379 RSC.
- K. Xue, Z. Liu, L. Jiang, D. Kai, Z. Li, X. Su and X. J. Loh, Biomater. Sci., 2020, 8, 926–936 RSC.
- I. Seah, X. Zhao, Q. Lin, Z. Liu, S. Z. Z. Su, Y. S. Yuen, W. Hunziker, G. Lingam, X. J. Loh and X. Su, Eye, 2020, 1–16 Search PubMed.
- X. Liu, Z. Li, X. J. Loh, K. Chen, Z. Li and Y. L. Wu, Macromol. Rapid Commun., 2019, 40, 1800117 CrossRef.
- X. J. Loh, P. L. Chee and C. Owh, Small Methods, 2019, 3, 1800313 Search PubMed.
- A. Lapprand, F. Boisson, F. Delolme, F. Méchin and J. P. Pascault, Polym. Degrad. Stab., 2005, 90, 363–373 CrossRef CAS.
- Z. Wang, S. Gangarapu, J. Escorihuela, G. Fei, H. Zuilhof and H. Xia, J. Mater. Chem. A, 2019, 7, 15933–15943 RSC.
- R. P. Houghton and A. W. Mulvaney, J. Organomet. Chem., 1996, 518, 21–27 CrossRef CAS.
- H. Ni, H. A. Nash, J. G. Worden and M. D. Soucek, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1677–1688 CrossRef CAS.
- S. Ephraim, A. Woodward and R. Mesrobian, J. Am. Chem. Soc., 1958, 80, 1326–1328 CrossRef CAS.
- A. Oberth and R. Bruenner, Ind. Eng. Chem. Fundam., 1969, 8, 383–388 CrossRef CAS.
- L. Rand, B. Thir, S. Reegen and K. Frisch, J. Appl. Polym. Sci., 1965, 9, 1787–1795 CrossRef CAS.
- S. J. Moravek and R. F. Storey, J. Appl. Polym. Sci., 2008, 109, 3101–3107 CrossRef CAS.
- B. Jeong, S. W. Kim and Y. H. Bae, Adv. Drug Delivery Rev., 2002, 54, 37–51 CrossRef CAS.
- H. J. Moon, D. Y. Ko, M. H. Park, M. K. Joo and B. Jeong, Chem. Soc. Rev., 2012, 41, 4860–4883 RSC.
- K. Mortensen and Y. Talmon, Macromolecules, 1995, 28, 8829–8834 CrossRef CAS.
- K. Mortensen and J. S. Pedersen, Macromolecules, 1993, 26, 805–812 CrossRef CAS.
- K. Mortensen and W. Brown, Macromolecules, 1993, 26, 4128–4135 CrossRef CAS.
- T.-H. Kim, Y.-S. Han, J.-D. Jang and B.-S. Seong, J. Appl. Crystallogr., 2014, 47, 53–59 CrossRef CAS.
- K.-C. Shih, C.-Y. Li, W.-H. Li and H.-M. Lai, Soft Matter, 2014, 10, 7606–7614 RSC.
- S. Manet, A. Lecchi, M. Impéror-Clerc, V. Zholobenko, D. Durand, C. L. P. Oliveira, J. S. Pedersen, I. Grillo, F. Meneau and C. Rochas, J. Phys. Chem. B, 2011, 115, 11318–11329 CrossRef CAS.
- I. Grillo, I. Morfin and S. Prévost, Langmuir, 2018, 34, 13395–13408 CrossRef CAS.
- S. Sami, E. Yildirim, M. Yurtsever, E. Yurtsever, E. Yilgor, I. Yilgor and G. L. Wilkes, Polymer, 2014, 55, 4563–4576 CrossRef CAS.
- E. Yılgör, İ. Yılgör and E. Yurtsever, Polymer, 2002, 43, 6551–6559 CrossRef.
- T. Tanaka, T. Yokoyama and Y. Yamaguchi, J. Polym. Sci., Part A-1: Polym. Chem., 1968, 6, 2137–2152 CrossRef CAS.
- A. J. A. Aquino, D. Tunega, G. Haberhauer, M. H. Gerzabek and H. Lischka, J. Phys. Chem. A, 2002, 106, 1862–1871 CrossRef CAS.
- W. R. Fawcett, J. Phys. Chem., 1993, 97, 9540–9546 CrossRef CAS.
- B. J. Lommerts, Polymer, 2001, 42, 6283–6287 CrossRef CAS.
- B. Bahramian, Y. Ma, R. Rohanizadeh, W. Chrzanowski and F. Dehghani, Green Chem., 2016, 18, 3740–3748 RSC.
- R. H. Lambeth, S. J. Pederson, M. Baranoski and A. M. Rawlett, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5752–5757 CrossRef CAS.
- P. C. Rooney, Preparation of polyesters with tin catalyst, US Pat., US5166310A, 1991 Search PubMed.
- J. Cervantes, R. Zárraga and C. Salazar-Hernández, Appl. Organomet. Chem., 2012, 26, 157–163 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ma00734j |
| ‡ These dilute reaction conditions were chosen such that the resulting PUs do not contain excessive crosslinks by allophanate bond formation and molecular weights too high to prevent dissolution in water, which is a necessary condition for the PUs to be used as thermogels for biomedical applications. |
| § These solvents were chosen as they have boiling points above 110 °C. |
| This journal is © The Royal Society of Chemistry 2020 |