
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrospray-based synthesis of fluorescent poly(D,L-lactide-co-glycolide) nanoparticles for the efficient delivery of an anticancer drug and self-monitoring its effect in drug-resistant breast cancer cells†
Manosree
Chatterjee
ab,
Ritwik
Maity
 c,
Souvik
Das
d,
Nibedita
Mahata
b,
Biswarup
Basu
*d and
Nripen
Chanda
*a
c,
Souvik
Das
d,
Nibedita
Mahata
b,
Biswarup
Basu
*d and
Nripen
Chanda
*a
aMaterial Processing and Microsystem Laboratory, CSIR – Central Mechanical Engineering Research Institute, Durgapur-713209, India. E-mail: n_chanda@cmeri.res.in; Fax: +91-343-2546745; Tel: +91-8902506209
bDepartment of Biotechnology, National Institute of Technology Durgapur, Durgapur-713209, India
cDepartment of Biochemistry and Molecular and Cellular Biology, University of Zaragoza, 50009, Spain
dDepartment of Neuroendocrinology and Experimental Hematology, Chittaranjan National Cancer Institute, Kolkata-700026, India. E-mail: biswarup.basu@gmail.com; Tel: +91-8800883257
First published on 13th October 2020
Abstract
A novel approach used to synthesize antimetabolite-conjugated and intense blue fluorescence-emitting smart polymeric nanoparticles is reported for the efficient delivery of anticancer drugs and self-monitoring their effect in drug-resistant metastatic breast cancer cells. Metastatic breast cancer is the deadliest cancer in women as chemotherapy does not perform well in its treatment. To prepare the drug-loaded fluorescent nanoparticles, the FDA-approved non-fluorescent poly(D,L-lactide-co-glycolide) (PLGA) polymer was modified into a newly designed fluorescent PLGA polymer by the covalent conjugation of the biocompatible fluorophore 1-pyrenebutyric acid (PBA). The fluorescent PLGA–PBA polymer was then electrosprayed by applying a potential of 8.0 kV to synthesize mono-dispersed spherical fluorescent nanoparticles (size ∼40 nm). The surface of the PLGA–PBA nanoparticles was conjugated with the potent anticancer drug molecule methotrexate (MTX) through a linker molecule, ethylenediamine (EDA), to kill cancer cells. The fluorescence, FTIR, NMR, and mass spectroscopy results of PLGA–PBA and PLGA–PBA@MTX nanoparticles provided proof of the successful synthesis of PBA- and MTX-conjugated nanoparticles with stable fluorescence for monitoring the in vitro therapeutic effect. A significant internalization of the PLGA–PBA@MTX nanoparticles was observed inside MTX-resistant MDA-MB-231 and MCF7 cells predominantly via micropinocytosis. Since MTX is an analog of folic acid and encourages cell internalization through the FRα receptor, a higher population of PLGA–PBA@MTX nanoparticles was observed in MDA-MB-231 cells with higher cell cytotoxicity than in MCF-7 cells. The amide bond, which links the MTX molecules to the surface of the fluorescent PLGA–PBA nanoparticles, was found to be sensitive to acidic pH. The controlled release of MTX occurred at pH 6.0 for ∼6 days due to the acid-catalyzed amide bond hydrolysis. The lower IC50 value justified a high MTX loading (36%) with significant conjugation efficiency (91.4%), and a rapid drug release in acidic medium compared to that of the free MTX molecule and high apoptosis in the nanoparticle-treated cells were observed. This study presents the usefulness of an engineered nano-formulation in pH-sensitive drug release and tracking of the therapeutic response, which may provide potential benefit in breast cancer treatment.
1. Introduction
Breast cancer is the second leading cause of cancer-related death worldwide in women and 6–10% of breast cancer initially diagnosed is in the metastatic state.1,2 Several new anticancer drugs (trastuzumab, apatinib, pertuzumab, neratinib, and olaparib) have been developed during the last few decades to improve the therapeutic index of the chemotherapy regimen.3,4 However, the difficulty in the therapeutic use of these drugs is their poor bioavailability and rapid elimination from circulation, resulting in low accumulation of the drug at the desired location, which imposes a high dose requirement. Moreover, the development of drug-resistant behaviour in cancer cells and the non-specific distribution of the drugs leading to adverse side effects drastically reduce the therapeutic efficacy.5 To overcome these limitations, polymeric nanoparticles were developed as imperative drug delivery vehicles due to their excellent physicochemical properties like (a) high drug encapsulation efficiency, (b) protection of drug molecules from premature degradation, (c) sustained release of drugs at the desired location, (d) increased bioavailability by prolonging the blood circulation time, and (e) passive accumulation at the tumor site by the enhanced permeability and retention (EPR) effect.6 Indeed, the biocompatible nature, stability, and tunable drug release ability in the physiological environment made the polymer-based nanoparticles attractive as a preferred drug carrier among all types of US Food and Drug Administration (FDA)-approved particle-based drug delivery systems.7The presence of an imaging agent along with a deliverable therapeutic agent is useful to monitor the specificity and therapeutic progress. Loading of both agents in molecular forms within a single polymeric nanocarrier is challenging, as it may influence the photostability of the imaging molecules and have significant impacts on the pharmacokinetic behaviour of the drug delivery systems.8 Since the drug molecules have to be released from the carrier, it is necessary to integrate the imaging molecule as an intrinsic part of the polymer so that the properties and purpose of both agents prevail. This type of modification is more familiar in metallic nanoparticles, especially transition metal nanoparticles, due to their inherent chemical properties. In contrast, most of the polymer-based nanoparticles are difficult to modify as they lack the desired number of suitable functional groups.
The majority of polymeric nanoparticles are fabricated using biocompatible polymers to reduce undesirable systemic toxicity from the drug transporter.9 Among them, the PLGA polymer has attracted significant interest as an efficient drug delivery nanosystem due to the presence of hydroxyl and carboxyl end groups on both sides of the polymer chain along with an ester-bonded backbone of lactic acid and glycolic acid monomer units, which can be used for stable conjugation of the imaging agents as well as the drug molecules.10 Yoo et al. conjugated drug-cum-imaging doxorubicin molecules with the p-nitro phenyl chloroformate-activated terminal hydroxyl group of PLGA to introduce a 10-fold more cytotoxic effect in HepG2 cells than free doxorubicin after 72 h.11 Liu et al. developed biodegradable PLGA nanoparticles, which were conjugated with an imaging molecule, rhodamine B, and a targeting agent, folic acid, at the copolymer end by the esterification reaction for target-specific imaging application.12 C. W. Liu and W. J. Lin reported the conjugation of PEG-bis-amine with PLGA followed by epidermal growth factor (EGF) receptor-targeting NR7 peptide and FITC molecules to synthesize doxorubicin encapsulated nanoparticles. They observed a faster pH-dependent in vitro doxorubicin release but stable FITC linkage to monitor the therapeutic response in SKOV3 cells.13 Thus, conjugation of imaging and therapeutic agents in a single PLGA platform is becoming an essential method that could be beneficial to execute multiple tasks at a time for better theranostic performance.
Inspired by the above example, we proposed a novel synthesis of 1-pyrenebutyric acid (PBA)-conjugated PLGA polymer using ethylenediamine as a linker molecule to fabricate a stable fluorescent nanoparticle with strong blue emission. The covalent conjugation of the PBA molecule with PLGA polymer would reduce the risk of premature loss of the fluorescent molecules and stabilize the fluorescence intensity at the imaging site. PBA is a biocompatible fluorophore, used in various bioimaging applications, as reported elsewhere.14,15 One of the examples reported by Wang et al. is the synthesis of 1-pyrenebutyric acid (PBAC)/β-cyclodextrin (β-CD) fluorescent nanoparticles for the fluorometric quantification of calf thymus DNA with a detection limit of 0.01 μg mL−1.14
Another objective of the present work is to load suitable drug molecules into the system so that the drug can be released to attain an effective drug concentration in drug-resistant metastatic breast cancer cells at a low therapeutic dose. To achieve this goal, an antimetabolite of folic acid, methotrexate (MTX), is applied to conjugate on the surface of the synthesized fluorescent PLGA–PBA nanoparticles. This potent anticancer molecule specifically interacts with reduced folate carrier (RFC) and, to some extent, with folate receptor α (FRα), which is overexpressed on the basolateral surface of cancerous tissues such as breast, ovarian and lung carcinoma.16,17 Despite revolutionary discoveries, cancer treatment still faces some challenges, including the variable expression of the tumor-specific receptors within a particular tumor tissue, which leads to tumor heterogeneity and the gradual development of resistance to the drugs, a frequently observed phenomenon in breast cancer.18,19 This problem can be resolved if the drug encounters the cancer cells in multiple ways and internalizes inside the cell by both cell-surface biomarker-dependent and independent pathways, which warrants investigation. If internalization happens through both pathways, there is the possibility of an increased uptake of nanoparticles inside the cancer cells that could enhance the therapeutic effect.
Methotrexate can play a dual role—as an anti-proliferative drug and a as targeting agent, as described by P. T. Wong and S. K. Choi in their report on folate receptor-positive KB tumor cells.20 The recent findings of Nogueira et al. regarding the conjugation pattern of the MTX molecule demonstrated that the carboxylic terminal of the MTX molecule is engaged in conjugation, leaving the amino-terminal free for receptor binding; as a result, the internalization of MTX-conjugates into epithelial cancer cells through FAα is accelerated.21 Once the targeted nanoparticles internalized through endocytosis, the conjugated drug molecules were released from the nanoparticles inside the cell due to the acidic pH of the lysosome. The released MTX inhibits nucleotide biosynthesis by irreversibly binding with the key enzymes of the synthesis pathways.17,22 The nucleotide deficiency leads to DNA damage by disrupting DNA synthesis and eventually, cell death.23 We were encouraged by their study to design a PLGA nanoparticle by making use of the MTX molecule as a drug and FRα receptor-specific agent to study its internalization and therapeutic efficacy in the metastatic breast cancer (MBC) cell line.
In this present study, a novel PBA-conjugated PLGA polymer was synthesized to develop fluorescent nanoparticles using an electrospray method. The PLGA–PBA nanoparticles were then covalently conjugated with MTX molecules through a simple surface functionalization approach using the ethylenediamine-mediated aminolysis reaction (Scheme 1). The efficiency as a drug delivery vehicle and self-monitoring agent of the designed nanoparticles was explored by studying the conjugation efficiency, pH-dependent drug release kinetics, and therapeutic efficacy on MTX resistant metastatic breast cancer cells.
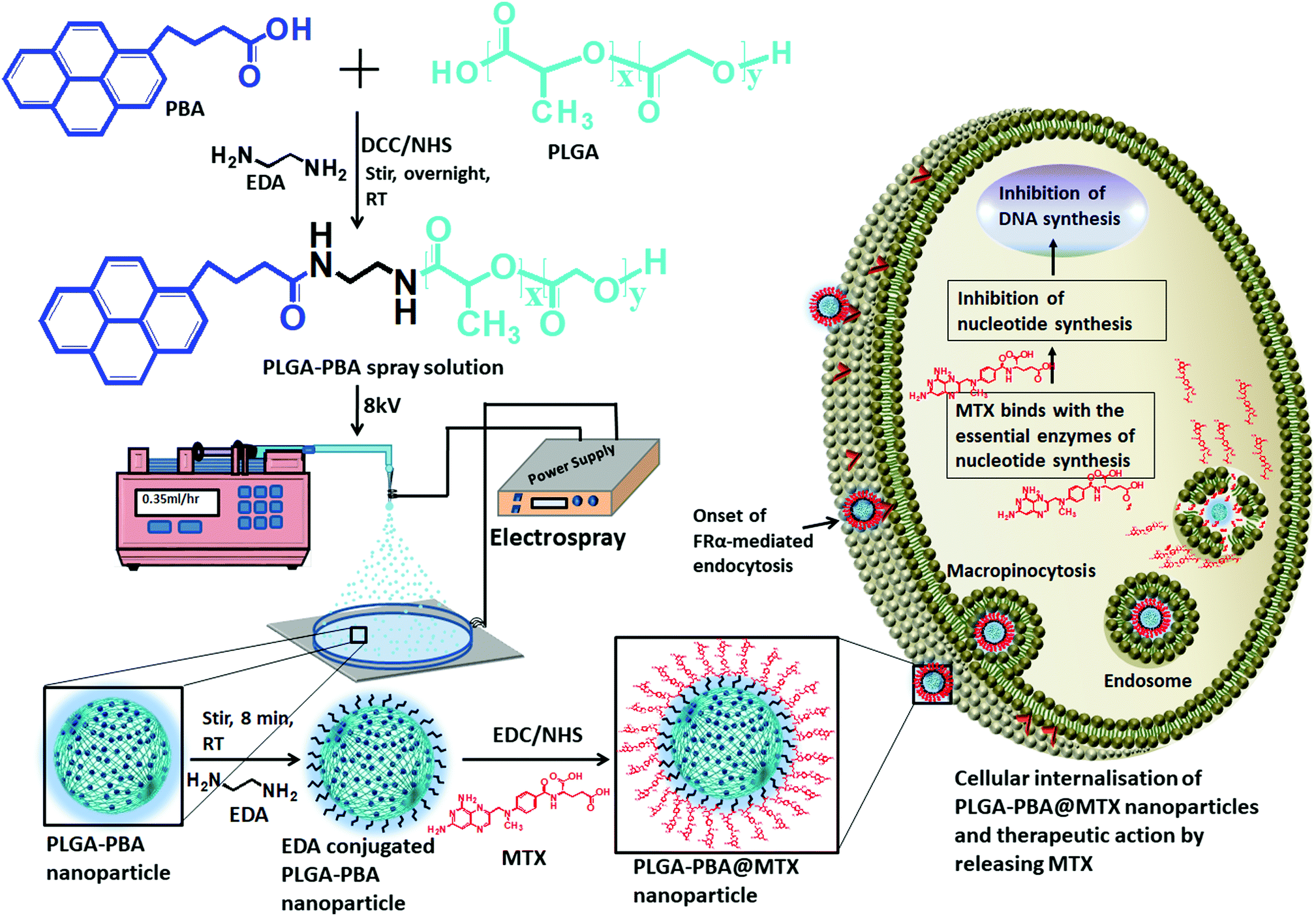 | ||
| Scheme 1 Schematic representation of the PLGA–PBA nanoparticle synthesis from the PBA-modified PLGA polymer and the subsequent preparation of PLGA–PBA@MTX nanoparticles to demonstrate the therapeutic action by releasing MTX after intracellular delivery. | ||
2. Experimental section
2.1 Materials
Poly(D,L-lactide-co-glycolide) (PLGA, acid terminated Mw 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–38000 with 50:50 lactide:glycolide ratio), methotrexate hydrate (MTX), 1-pyrenebutyric acid (PBA), dialysis kit (Pur-A-LyzerTM Maxi 12000), paraformaldehyde (reagent grade, crystalline), (N-ethyl-N′-(3-(dimethylamino)propyl)) carbodiimide (EDC), N-hydroxysuccinimide (NHS), dicyclohexyl carbodiimide (DCC) and phosphate buffer solution (10×, pH 7.4, pH 4.0 and pH 9.0) were purchased from Merck, India. All other chemicals like acetone, dichloromethane (DCM), diethyl ether, HPLC (High-performance liquid chromatography) grade water and sodium hydroxide were purchased from Merck, India. Human metastatic breast cancer cell lines (MCF-7 and MDA-MB-231) were obtained from NCCS (National Center for Cell Sciences) Pune, India and cultured according to the instructed protocol. The reagents used in the cellular efficiency study like Dulbecco's modified Eagle medium (DMEM), penicillin–streptomycin (10000 U mL−1) (antibiotic) solution, fetal bovine serum (FBS, certified, heat-inactivated, US origin), phosphate buffer saline (PBS, pH 7.4), MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) and ProLong Diamond Antifade Mountant were purchased from Gibco (Life Technologies). The reagents used for cellular internalization pathway blocking study like lovastatin (CAS Number 75330-75-5), chlorpromazine hydrochloride (CAS Number 69-09-0), miloride hydrochloride (CAS Number 2016-88-8) and acridine orange (CAS Number 10127-02-3) were purchased from Merck, India. The annexin V Alexa Fluor 488/propidium iodide staining kit was obtained from Invitrogen, USA.
000–38000 with 50:50 lactide:glycolide ratio), methotrexate hydrate (MTX), 1-pyrenebutyric acid (PBA), dialysis kit (Pur-A-LyzerTM Maxi 12000), paraformaldehyde (reagent grade, crystalline), (N-ethyl-N′-(3-(dimethylamino)propyl)) carbodiimide (EDC), N-hydroxysuccinimide (NHS), dicyclohexyl carbodiimide (DCC) and phosphate buffer solution (10×, pH 7.4, pH 4.0 and pH 9.0) were purchased from Merck, India. All other chemicals like acetone, dichloromethane (DCM), diethyl ether, HPLC (High-performance liquid chromatography) grade water and sodium hydroxide were purchased from Merck, India. Human metastatic breast cancer cell lines (MCF-7 and MDA-MB-231) were obtained from NCCS (National Center for Cell Sciences) Pune, India and cultured according to the instructed protocol. The reagents used in the cellular efficiency study like Dulbecco's modified Eagle medium (DMEM), penicillin–streptomycin (10000 U mL−1) (antibiotic) solution, fetal bovine serum (FBS, certified, heat-inactivated, US origin), phosphate buffer saline (PBS, pH 7.4), MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) and ProLong Diamond Antifade Mountant were purchased from Gibco (Life Technologies). The reagents used for cellular internalization pathway blocking study like lovastatin (CAS Number 75330-75-5), chlorpromazine hydrochloride (CAS Number 69-09-0), miloride hydrochloride (CAS Number 2016-88-8) and acridine orange (CAS Number 10127-02-3) were purchased from Merck, India. The annexin V Alexa Fluor 488/propidium iodide staining kit was obtained from Invitrogen, USA.
2.2 General characterization procedure
All the steps for the synthesis of PLGA–PBA@MTX nanoparticles were characterized by fluorescence spectroscopy (Agilent Technologies, Cary Eclipse) to identify whether the conjugation of the PBA molecule was successful and stable. The hydrodynamic size and charge of the fluorescent PLGA nanoparticles before and after MTX conjugation (PLGA–PBA and PLGA–PBA@MTX) were measured using a particle size analyzer (Nano Sight NS500). The morphology of the PLGA–PBA nanoparticles and PLGA–PBA@MTX nanoparticles was studied using various microscopic techniques such as field emission scanning electron microscopy (FESEM, Model: Zeiss Sigma HD), atomic force microscopy (AFM, Model: Nano Surf C3000) and transmission electron microscopy (TEM, JEOL 3010). The molecular compositions of the PLGA–PBA polymer and PLGA–PBA@MTX nanoparticles were established by Fourier-transform infrared spectroscopy (FT-IR, Model: JASCO FT/IR-4700) and mass spectroscopy (micrOTOF-Q II 10330). CHN and EDX analyses were carried out to measure the percentages of carbon, hydrogen and nitrogen in the PLGA–PBA and PLGA–PBA@MTX nanoparticles. The 1H nuclear magnetic resonance (1H NMR, BRUKER) spectroscopic study was carried out in DMSO at 500 MHz for each step from PBA-conjugated polymer synthesis to PLGA–PBA@MTX nanoparticle fabrication. The calibration curve, drug loading and release profile of MTX were obtained by using UV-VIS spectroscopy (Agilent Technologies, Cary 60 UV-Vis) analysis.2.3 Synthesis of 1-pyrenebutyric acid-terminated PLGA polymer
The synthesis of PBA-terminated PLGA polymer (PLGA–PBA) was achieved in two steps, as reported by Kim et al., with some modifications.24 Briefly, in the first step, 1.0 g of PLGA was dissolved in 8.0 mL of DCM. Then, DCC/NHS (1:1) in 10 times molar excess with respect to PLGA was added to the PLGA solution and stirred for 3 hours (h), which activates the –COOH group of the PLGA polymer chain. EDA in DCM solution was added dropwise to the activated PLGA solution (molar ratio of PLGA:EDA was 1:2) under continuous stirring at room temperature. After overnight reaction, the amine-terminated PLGA polymer was precipitated and washed with ice-cold diethyl ether four times, and then dried under vacuum.
In the second step, the amine-terminated PLGA polymer was redissolved in DCM and then dropwise added into the DCC/NHS-activated PBA solution in DCM (molar ratio of PLGA:PBA was 1:5) under continuous stirring at room temperature. After overnight stirring, the PBA-conjugated PLGA polymer (PLGA–PBA) precipitated out and was washed with ice-cold diethyl ether four times. The PLGA–PBA polymer was also washed with acetone to remove the by-product, dicyclohexylurea, of the DCC reaction and dried under vacuum. Yield: 0.32 g (86.13%); fluorescence λex: 240 nm and λem: 378 nm and 398 nm. 1H NMR of PLGA polymer, δ (ppm) = 1.48 (3H, CH3), 4.89 (2H, CH2), 5.24 (1H, CH). 1H NMR of PLGA–PBA polymer, δ (ppm) = 1.48 (CH3 of PLGA), 2.09, 2.6, and 2.7 (CH2 of PBA), 4.89 (CH2 of PLGA), 5.24 (CH of PLGA), 5.57–5.59 (CH2 of EDA), 7.91–8.42 (CH of PBA), 10.56–10.61 (NH of EDA).
2.4 Synthesis of fluorescent PLGA–PBA nanoparticles
The PLGA–PBA polymer was used to fabricate the fluorescent PLGA–PBA nanoparticles by following our previously reported electrospray method.25 Briefly, the PLGA–PBA polymer was dissolved in acetone (3.0%, W/V) then stirred for 10 min to form a homogeneous solution. Following the previously reported electrospray method, the polymer solution was dribbled at a flow rate of 0.35 mL h−1 from a syringe pump through a conductive needle connected with a high voltage power supply. At 8.0 kV, the polymer droplet was sprayed on a grounded aqueous collecting medium, which was placed 10 cm below the needle point. The resultant nanoparticle (PLGA–PBA) suspension was stored at 4 °C for further use.2.5 Conjugation of methotrexate with PLGA–PBA nanoparticles
Methotrexate molecules were anchored on the surface of the PLGA–PBA nanoparticles through EDA linker molecules. EDA was first bonded to the PLGA–PBA nanoparticle by aminolysis of the ester linkage of the PLGA backbone. For the aminolysis, the electrospray synthesized PLGA–PBA nanoparticles were incubated with 5.0 mM EDA for 8 min and immediately centrifuged at 18000 rpm for 10 min at 15 °C to separate the excess EDA. The EDA-conjugated PLGA–PBA nanoparticle pellet was resuspended in HPLC water. On the other hand, a 7.0 mM aqueous MTX solution (pH ∼ 7.0) was activated by EDC/NHS (1:4) following the protocol reported by Dixit et al.26 The activated MTX was added to the EDA-conjugated PLGA–PBA nanoparticle suspension under continuous stirring for 5 h. This final conjugate was centrifuged at 18000 rpm for 10 min at 15 °C to separate the unreacted MTX if any. The MTX-conjugated PLGA–PBA nanoparticles (PLGA–PBA@MTX) were washed with HPLC water three times and stored at 4 °C. Yield: 3.3 mg (74.98%); UV-Vis λmax: 300 nm; fluorescence λex: 240 nm and λem: 378 nm and 398 nm. 1H NMR of PLGA@MTX nanoparticles, δ (ppm) = 1.48 (CH3 of PLGA), 3.21 (N-CH3 of MTX), 4.79 (CH2 of EDA and MTX), 4.89 (CH2 of PLGA), 5.24 (CH of PLGA), 6.63 (NH2 of MTX), 7.46–7.67 (NH of EDA), 7.67–8.2 (CH of aromatic pteridine ring and p-aminobenzoic acid), 8.57 (1CH of pteridine ring), 12.48–12.5 (COOH of MTX).
2.6 Stability study
Stability of the nanoparticles at physiological pH is essential to study their effect on the metastatic breast cancer cell lines (MCF-7 and MDA-MB-231). To check the stability, the sizes of the PLGA–PBA nanoparticles before and after MTX conjugation (PLGA–PBA and PLGA–PBA@MTX) were measured using a particle size analyzer after dispersing them in 1× PBS of different pH values (9.0, 7.4, 6.0, and 4.0) for 7 days. Besides, the stability of both nanoparticles was also checked by using fluorescence spectroscopy. For this purpose, the individual nanoparticle suspension in 1× PBS (pH 7.4) was placed into a dialysis tube with a molecular cut-off of 12000 Da. The dialysis tube was immersed in a pool of release medium (1× PBS pH 7.4) under a constant stirring (500 rpm) condition. At each time interval, a 1.0 mL aliquot of release medium was withdrawn and the fluorescence of the release medium was checked using a fluorescence spectrophotometer. After each measurement, the aliquot was returned back to the pool of the medium to maintain the dilution factor.
2.7 Determination of drug loading and conjugation efficiency
To measure the drug loading and conjugation efficiency of the nanoparticles, the initial and conjugated amounts of MTX were calculated from its calibration curve that depicts the relation between its absorbance and concentration. The calibration curve was determined by measuring known concentrations of MTX solution in HPLC water using UV-Vis spectroscopy and then plotting the absorbance versus concentration graph. To establish the drug loading and conjugation efficiency, 10 μL of the initially given MTX solution and the same amount of PLGA–PBA@MTX suspension were taken into two separate volumes of 1.0 mL of HPLC water and the absorbance was measured using UV-Vis spectroscopy. The amount of MTX was calculated from the calibration curve. The percentage drug loading and conjugation efficiency was calculated using equation 1 and 2, respectively.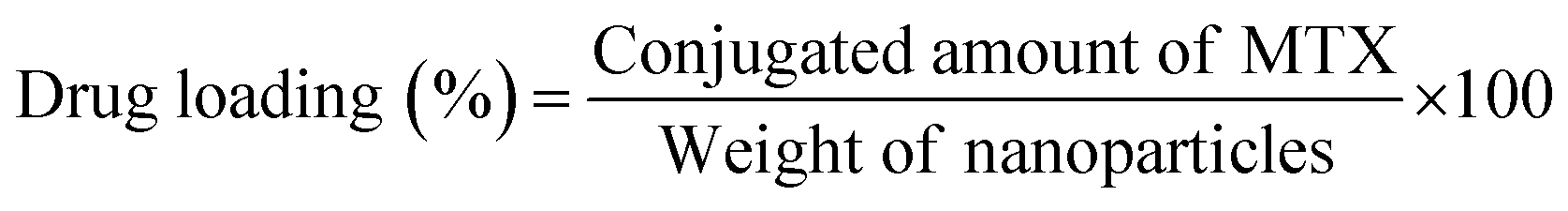 | (1) |
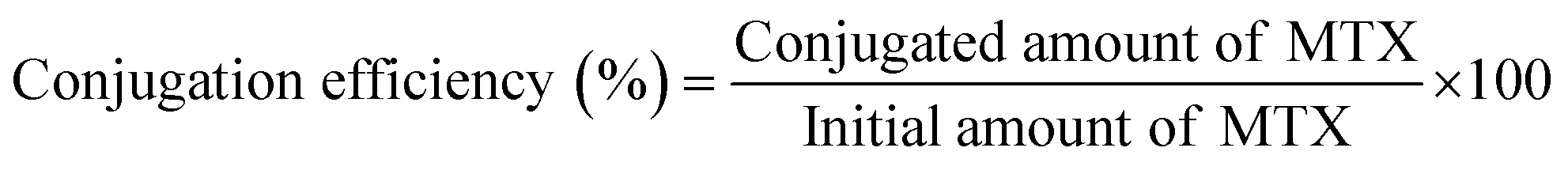 | (2) |
2.8 In vitro drug release kinetics
The release profile of the drug-conjugated nanoparticles in PBS buffer (1× PBS of pH 6.0 and pH 4.0) was monitored to explore the rate and pattern of the drug release kinetics of the PLGA–PBA@MTX nanoparticles. For this purpose, nanoparticles (56.0 μg) were suspended in 3.0 mL of 1× PBS (pH 6.0 and pH 4.0) and placed into dialysis tubes with a molecular cut-off of 12000 Da. The dialysis tubes were immersed in 8 times excess release medium (1× PBS of pH 6.0 and pH 4.0, respectively) under constant stirring (500 rpm). At a constant time interval, a 1.0 mL aliquot of release medium was withdrawn to check the amount of MTX released using UV-Vis spectroscopy and then returned to the pool of release medium to maintain the dilution factor. The absorbance of the release medium was correlated with the calibration curve and the amount of MTX released was determined from PLGA–PBA@MTX at a specific time point. After ∼96% MTX release, its percentage of release against a particular point of time was plotted to depict the kinetics of MTX release from PLGA–PBA@MTX. The best-fitting curve and the parameters for the release constant (K), release exponent (n) and regression coefficient (R2) were determined by fitting the experimental release data into the Korsmeyer–Peppas kinetic model.
The equation used in curve fitting is as follows:
| F = (Mt/M) = Kmtn | (3) |
In this equation, Mt/M is the fraction of drug released from PLGA–PBA@MTX at time t, K is the release rate constant and n is the release exponent.
2.9 Hemolysis test of PLGA–PBA@MTX
The hemolytic activity of PLGA–PBA@MTX was evaluated in anticoagulant-treated human blood taken from normal volunteers. 0.5 μM, 1.1 μM and 2.0 μM concentrations of PLGA–PBA@MTX were added to the whole blood and incubated for 2 h at 37 °C in an incubator. After centrifugation at 3000 rpm for 10 min, the release of hemoglobin was measured via a microplate reader at 540 nm.27 Phosphate buffer saline (pH 7.4) solution and distilled water have been used as a positive control and a negative control, respectively. The experiment was carried out in triplicate and less than 5.0% hemolysis was considered to be significant. The following formula was used for calculating the hemolysis rate.28| Hemolysis (%) = (ODSample − ODNegative)/(ODPositive − ODNegative) × 100 | (4) |
2.10 Cytotoxicity study
MTT assay was performed for different concentrations of PLGA–PBA@MTX nanoparticles to investigate the cytotoxicity in MDA-MB-231 and MCF-7 cell lines (1× 104 cells per mL; 37 °C). Cells at the exponential growth phase were seeded in polystyrene-coated 96-well plates and incubated for 24 h at 37 °C. Then, the cells were treated with different concentrations of MTX in PLGA–PBA@MTX ranging from 15 nM to 2.6 μM for MDA-MB-231 and from 5 μM to 80 μM for MCF-7 and the equivalent concentration of free MTX for the respective cell lines. After 48 h incubation, 10 μL of MTT per well (5 mg mL−1, a stock solution in 1× PBS) was added and left for 4 h. Then, the formed formazan crystals were dissolved in 100 μL of DMSO for 30 min in the dark at 37 °C and the intensity of the developed color was measured using a plate reader. Untreated cells were considered 100% viable. The obtained intensities were represented as dosage vs viability response charts. The absorbance was measured on an ELISA plate reader (iMark™ Microplate Absorbance Reader) at a test wavelength of 570 nm and a reference wavelength of 650 nm.2.11 Pathway blocking study
The cellular internalization mechanism was studied using a previously described method by our group.28 For this study, a non-toxic concentration of MTX (520 nM for MDA-MB-231 and 13 μM for MCF-7), which was present in PLGA–PBA@MTX nanoparticles, was incubated with: (a) cells at 4 °C and 37 °C without any inhibitors; (b) cells in the presence of a clathrin-mediated endocytosis blocker; (c) cells in the presence of a caveolae-mediated endocytosis blocker and (d) cells in the presence of a macro-pinocytosis blocker. For this purpose, MDA-MB-231 and MCF-7 cells were grown on poly-L-lysine-treated coverslips and treated with 50 μM chlorpromazine (affecting the clathrin lattice assembly), 50 μM lovastatin (blocking lipid raft-mediated endocytosis via cholesterol depletion) and 100 μM amiloride (blocking macro-pinocytosis via sub-membranous acidification).28–32 After treatment with the blockers, cells were incubated with PLGA–PBA@MTX for 2 h at 37 °C. For the energy-dependent study, cells were incubated for 30 min at 4 °C followed by nanoparticle treatment for 2 h at 4 °C. Post treatment, cells were washed to remove unbound PLGA–PBA@MTX and microscopic slides were prepared. Images were then captured by an EVOS fluorescence microscope, and the fluorescence intensities of 20 cells from different fields were calculated using the ImageJ processing package (NIH, USA). The results were presented as mean ± SD from three independent experiments.2.12 Endosome staining with acridine orange
To confirm the presence of PLGA–PBA@MTX nanoparticles inside the cellular endosome, acridine orange staining was performed using a previously described method.33 Briefly, cells were cultured on poly-L-lysine-coated slides and treated with a non-toxic concentration of PLGA–PBA@MTX nanoparticles for 2 h. Then, the slides were washed and stained with acridine orange (5 μg mL−1) for 5 min. After thorough washing, the cells were visualized under an EVOS fluorescence microscope using red and blue fluorescence emission filters for the endosomes and PLGA–PBA@MTX nanoparticles, respectively, to study the localization of the nanoparticles inside the cells. Acridine Orange was used to locate the cell's nucleus as it emits green fluorescence when bound to double stranded DNA.2.13 Annexin V Alexa Fluor 488/propidium iodide staining
To explore PLGA–PBA@MTX nanoparticle-induced apoptosis, MDA-MB-231 and MCF-7 cells were grown overnight and incubated with three concentrations of PLGA–PBA@MTX nanoparticles (with respect to MTX concentration) and equivalent concentrations of free MTX for 24 h. Cells were stained in the dark with Alexa Fluor 488-conjugated Annexin V (1 μg mL−1) and PI (0.2 μg mL−1) in a Ca2+-enriched binding buffer for 30 min at room temperature and then analyzed by a two-color flow cytometric assay. Invitrogen Alexa Fluor 488 dye is a bright, green-fluorescent dye (emission max at 525 nm) with excitation ideally suited to the 488 nm laser line. Annexin V and PI emissions were detected using FITC and PI filters, respectively, in a flow cytometer system (Becton Dickinson FACSVerse). The percentage apoptosis was quantified from the mean ± SD of at least three independent experiments.2.14 Comet assay
The comet assay was performed following the procedure reported by Nandhakumar et al.34 In brief, MDA-MB-231 cells were cultured on a 35 mm plate overnight; then, the cells were treated: one set with PLGA–PBA@MTX nanoparticles (MTX concentration: 1.1 μM), another with an equivalent concentration of free MTX, and one set remained untreated. Then, the cells were collected and washed two times with PBS; then, they were suspended in 100 μL of 1% LMPA (low melting point agarose). 50 μL of cell suspension from each group was taken and poured as a smear with the help of a coverslip on a comet slide precoated with 1% NMA (normal melting agarose) (base coating) and placed on a chilled ice pack for 1 h. Once the cell-LMPA layer solidified, another 75 μL of 0.5% LMPA was added on the slide as a smear and allowed to solidify. The slides were carefully immersed in lysis solution (2.5 M NaCl, 100 mM EDTA, 10 mM Tris–HCl, 1% Triton X-100, 10% DMSO, and pH 10) overnight at 4 °C. After lysis, the slides were placed in a gel-tank filled with alkaline electrophoresis buffer (300 mM NaOH, 1 mM EDTA, and pH 13) and kept for 30 min to unwind the DNA followed by electrophoresis at 300 mA for 45 min. After electrophoresis, the slides were neutralized in 0.4 mM Tris–HCL and air-dried. The slides were stained using 1 mg mL−1 EtBr solution. Excess stain was washed with distilled water and the slides were viewed under a fluorescence microscope at 100× magnification with an excitation filter of 560 nm. The OpenComet v1.3.1 software was used to analyze the comet assay images.2.15 Statistical analysis
Data were expressed as mean ± standard deviation. Results were analyzed by ANOVA followed by Tukey's post-test. The statistical analysis was performed using Graph Pad Prism 7.0 software (GraphPad Software Inc., La Jolla, CA, USA).3. Results and discussion
3.1 Modification of PLGA polymer and preparation of PLGA–PBA@MTX nanoparticles
The modification of PLGA polymer with the fluorescent PBA molecule and its use in the synthesis of PLGA–PBA@MTX nanoparticles to enable therapeutic action towards the cancer cells are illustrated in Scheme 1. During the synthesis, it was observed that PBA molecules lose their fluorescence properties in the presence of MTX if they are mixed at the beginning (as shown in Fig. S2, ESI†). The reason for this quenching of fluorescence may be due to energy transfer from PBA to MTX molecules after excitation.35 To retain the fluorescence properties in the nanoparticles, PBA was first covalently conjugated with the PLGA molecules to make it an intrinsic part of the polymer. The off-white color compound obtained from this step showed similar fluorescence properties to those of free PBA in acetone, suggesting the formation of PBA-terminated PLGA polymer. In the second step, PLGA–PBA in acetone was electrosparyed through a conductive needle at a flow rate of 0.35 mL h−1 and an applied voltage of 8.0 kV. At this flow rate, the PLGA–PBA solution forms spherical droplets, which eventually deform under the high potential difference. In this process, as soon as the electric field intensity reached the Rayleigh limit, a Taylor cone is formed, which undergoes Coulomb fission leading to the formation of stable PLGA–PBA nanoparticles.25 The third or last step deals with the EDA-assisted functionalization of PLGA–PBA nanoparticles with MTX molecules using aminolysis chemistry as reported in our previous publication (Fig. 1). In one of our earlier studies, we successfully functionalized the PLGA pattern surface with anti-EpCAM (epithelial cell adhesion molecule) antibody using EDA by aminolysis of the PLGA nanostructure.36 Likewise, we enriched the surface of the PLGA–PBA nanoparticles with –NH2 groups by the EDA-assisted aminolysis of the ester bond of the PLGA–PBA chain. It was assumed that only the carbonyl groups of the PLGA–PBA polymer chain, which cover the surface of the nanoparticles, are dissociated by the nucleophilic attack of the EDA molecules after a short exposure time.37 This aminolysis generates –NH2 groups on the surfaces of PLGA–PBA nanoparticles, which were used in the formation of amide bonds with EDC/NHS activated –COOH groups of the MTX molecules. In this way, the amine groups of the MTX molecules remain free for interaction with FAα receptors of the proposed cancer cells at the time of receptor-mediated internalization of the PLGA–PBA@MTX nanoparticles. | ||
| Fig. 1 Synthesis of PLGA–PBA@MTX nanoparticles. | ||
3.2 Characterization
The synthesis of PLGA–PBA@MTX nanoparticles involves step-wise chemical conjugation of fluorescent PBA with PLGA polymer followed by conjugation of the MTX drug molecule with PLGA–PBA nanoparticles in the presence of ethylenediamine (EDA) and the coupling agents DCC/NHS and EDC/NHS, respectively (Fig. 1). Each step was followed using different characterization techniques like FTIR, NMR, mass, hydrodynamic size and surface charge measurements.The average hydrodynamic sizes of the PLGA–PBA nanoparticles before and after MTX conjugation were 71.0 nm and 252.1 nm, respectively (Table S1, ESI†). The size of the nanoparticles increases due to the conjugation of a number of MTX molecules on the surface of the PLGA–PBA nanoparticles. The zeta potential of the PLGA–PBA nanoparticles increased from −13.7 mV to −38.5 mV after conjugation with MTX due to the protrusion of electron-rich –NH2/–COOH groups of MTX molecules as well as the –OH groups of the aminolyzed PLGA molecules on the surface. A spherical morphology with a smooth particle surface and uniform size was observed through AFM, FESEM and TEM analyses for both the conjugated and unconjugated types of nanoparticles (Fig. 2). The average size of the PLGA–PBA nanoparticles and PLGA–PBA@MTX nanoparticles was ∼40 nm and ∼105 nm, respectively, as estimated from the size distribution graphs, which were obtained using ImageJ software (Fig. 2 insets).38 The smaller core-size of the PLGA–PBA@MTX nanoparticles could be favourable for passive targeting to the reduced folate carrier (RFC) and folate receptor α (FAα)-negative and methotrexate resistant cells.22
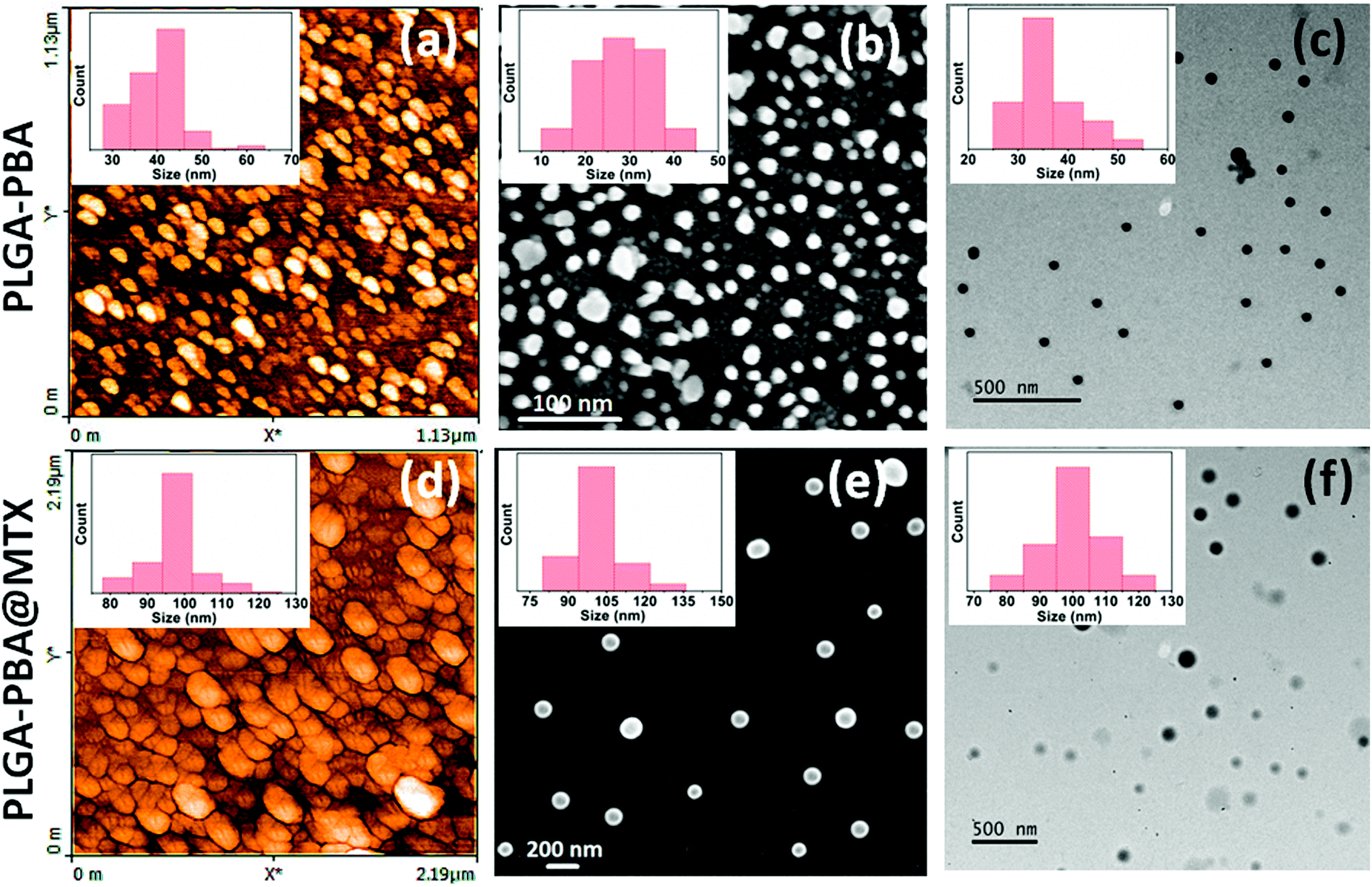 | ||
| Fig. 2 (a) AFM, (b) FESEM and (c) TEM images of PLGA–PBA nanoparticles and the same for PLGA–PBA@MTX nanoparticles are shown in (d), (e) and (f), respectively. The insets show the corresponding particle size distribution histograms. | ||
The fluorescence spectrum of the PBA molecule exhibited three emission peaks at 376 nm, 396 nm and 418 nm when excited at 240 nm (Fig. S1, ESI†).39 These characteristic fluorescence emission peaks of PBA remain unchanged after conjugation with PLGA polymer in the PLGA–PBA nanoparticles and PLGA–PBA@MTX nanoparticles when excited at 240 nm (Fig. 3a). This result indicates that PBA molecules retained their molecular properties in the PLGA–PBA nanoparticles as well as PLGA–PBA@MTX nanoparticles.
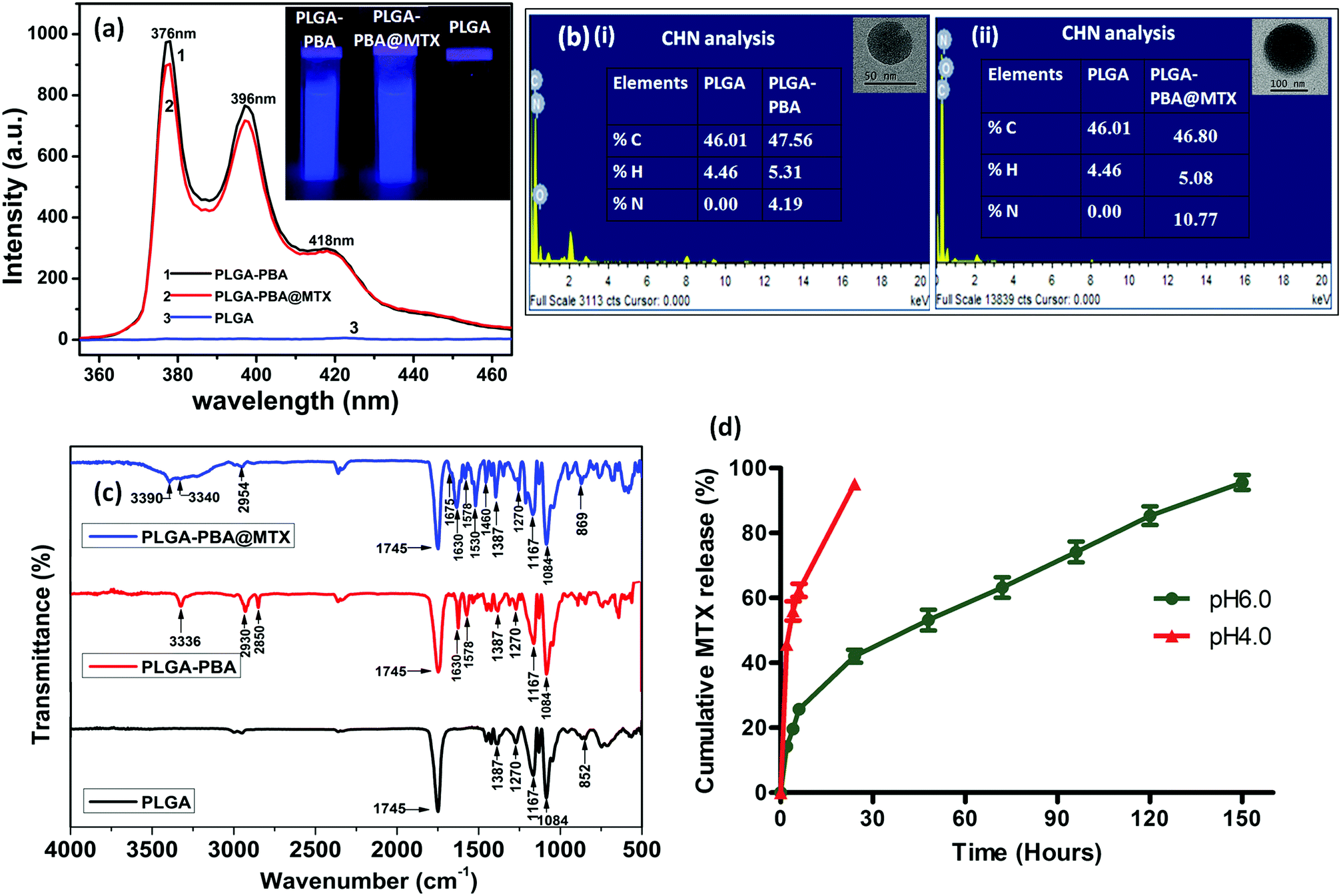 | ||
| Fig. 3 (a) Fluorescence spectra of PLGA, PLGA–PBA nanoparticles and PLGA–PBA@MTX nanoparticles. The images of nanoparticle suspensions under a UV lamp (λmax = 265 nm) are represented in the inset. (b) The EDX and CHN analyses of (i) PLGA–PBA nanoparticles and (ii) PLGA–PBA@MTX nanoparticles; their corresponding single nanoparticle images are presented in the insets. (c) FTIR spectra of PLGA polymer, PLGA–PBA polymer and PLGA–PBA@MTX nanoparticles showing chemical conjugation of PBA and MTX with PLGA before and after nanoparticle formation. (d) MTX release profiles of the PLGA–PBA@MTX nanoparticles at pH 6.0 and pH 4.0. | ||
In order to confirm the successful conjugation of MTX molecules, the percentage values of fundamental elements present in the pristine PLGA polymer and the subsequent PLGA–PBA and PLGA–PBA@MTX nanoparticles were obtained using CHN and EDX analysis techniques. The insets of Fig. 3b show the results of CHN analysis, indicating the increasing percentage of nitrogen from PLGA–PBA nanoparticles to PLGA–PBA@MTX nanoparticles. This result is also supported by EDX analysis of a single particle, as shown in Fig. 3b.
FTIR studies further supported the stepwise conjugation of PBA and MTX with PLGA before and after nanoparticle formation, respectively. PLGA polymer showed a strong carbonyl stretching frequency at 1745 cm−1 due to the ester bond between lactic acid and glycolic acid monomers, while weak peaks were observed at 2995–2850 cm−1 for the alkyl C–H group and at 1270–1087 cm−1 for the ether group, which were also observed in the spectrum of the PLGA–PBA polymer (Fig. 3c).40 The intense peak for the carbonyl group of PBA–COOH at 1686 cm−1 was shifted to 1630 cm−1 after amide linkage with –NH2 of EDA conjugated PLGA polymer (Fig. S3, ESI†).41 Apart from these carbonyl peaks, the peaks for N–H bending (amide II) and N–H stretching vibration appeared at 1578 cm−1 and ∼3340 cm−1, respectively, for the PLGA–PBA polymer before and after nanoparticle formation (Fig. 3c).42,43 The conjugation of MTX molecules with the PLGA–PBA nanoparticles was established from the signature peaks of the amide bond at 1630 cm−1 (amide I), 1578 cm−1 (amide II) and ∼3340 cm−1 (amide N–H stretch), which proved the successful synthesis of PLGA–PBA@MTX nanoparticles through amide linkage (Fig. 3c). The structure of the MTX molecule bears two amino groups, an aromatic pteridine ring and a p-aminobenzoic acid moiety with characteristic stretching vibrations at 3390 cm−1, 1640 cm−1 and 1678–1488 cm−1, which were also observed in the FTIR spectrum of the final nanoparticles with a slight shifting of the stretching frequencies (Fig. S3, ESI†).44 The study indicates that the two amino groups of the MTX molecules remain intact and free after conjugation, which is required for the binding with FRα on the cell surface. It is also worth noting that the characteristic absorption bands of the PLGA chain were distinctly observed among the distinctive absorption peaks of MTX and PBA in the FTIR spectrum of PLGA–PBA@MTX, which is expected as PLGA forms the backbone of the final nanoparticles.
1H NMR spectroscopic analysis, as shown in Fig. S4 (ESI†), further confirmed the FTIR results, which established the successful conjugation of PBA and MTX with PLGA molecules in the nanoparticles. The 1H NMR spectrum of the bare PLGA polymer showed chemical shifts (δ) at 1.48 ppm and 5.24 ppm for the CH3 and CH groups of lactic acid and 4.89 ppm for the CH2 group of glycolic acid, as reported by Wang et al.45 After PBA conjugation, peaks appeared at 7.91–8.42 ppm due to the aromatic protons of the pyrene group.46 Indeed, the PLGA–PBA polymer showed all the respective aliphatic and aromatic proton peaks of PLGA polymer and PBA molecules. The peaks for CH2 of –NHCH2– at 5.57–5.59 ppm were attributed to the EDA molecule in the PLGA–PBA polymer, which was used for the chemical conjugation between PBA and PLGA. To ensure the presence of the MTX molecule, only the PLGA nanoparticle was selected for MTX attachment, instead of the PLGA–PBA nanoparticle, as this helped to analyze the MTX conjugation through a simplified 1H NMR spectrum. In the 1H NMR spectrum of PLGA@MTX nanoparticles, peaks at 7.72–8.57 ppm corresponded to the protons of the aromatic pteridine ring and p-aminobenzoic acid moiety of the MTX molecule. A similar 1HNMR spectrum of MTX, which was conjugated with polyamidoamine dendrimer, was reported by Zhang et al.47 The peak assigned for –CONH– appeared at 7.46–7.67 ppm, which proved the formation of amide bonds between MTX and PLGA nanoparticles through EDA (Fig. S4, ESI†).47 During the conjugation, one of the –COOH groups of MTX participated in the amide bond formation with the PLGA-EDA conjugate leaving the other –COOH and –NH2 groups free, which produced peaks at 12.48–12.5 and 6.63 ppm, respectively. Thus, the 1H NMR results corroborated the MTX conjugation through the EDA molecule on the surface of the PLGA nanoparticles.
The FTIR and 1H NMR results were further strengthened by the mass spectroscopy analysis of PLGA–PBA polymer and PLGA–PBA@MTX nanoparticles, as shown in Fig. S5 and S6 (ESI†). The electrospray ionization of PLGA–PBA polymer displayed a major peak of [PBA-EDA–(LA)1–(GA)1]+ at 475.32 m/z (calculated value of 476.23). This peak value closely matched with the calculated mass of covalently linked PBA with the fragmented component of a PLGA molecule, i.e. one lactic acid (LA) and one glycolic acid (GA) through EDA, which was produced during the ionization process of analysis. The observed mass peaks of the PLGA–PBA polymer were similar with the calculated values of fragments like [PBA–EDA]+, [PBA–EDA–(LA)2–(GA)2]+ and [PBA–EDA–(LA)3–(GA)2]+ ions at 346.32, 606.17 and 679.51 m/z, (calculated values of 346.2, 606.26 and 678.28), respectively (Fig. S5, ESI†). Mass spectroscopy analysis after MTX conjugation with PLGA–PBA nanoparticles produced an intense peak at 455.1 m/z for [MTX]+ (calculated value at 454.44). This intense peak at 455.1 m/z may be due to the MTX molecules, which were conjugated on the surface of PLGA–PBA nanoparticles and separated at the time of electrospray ionization. The other peaks of ionized fractions of [MTX–EDA]+, [MTX–EDA–(LA)1]+, [MTX–EDA–(GA)2–(LA)1]+, [PBA–EDA]+, [PBA–EDA–(LA)1–(GA)1]+ and [PBA–EDA–(LA)3–(GA)2]+ were at 497.23, 568.56, 685.44, 346.32, 475.32 and 679.51 m/z, respectively (calculated values of 495.2, 567.26, 683.2, 346.2, 476.23 and 678.28) (Fig. S6, ESI†), confirming the presence of PBA and MTX in the PLGA networks through EDA-mediated chemical conjugation.48,49
3.3 Drug loading and conjugation efficiency
The amide bond that linked MTX on the surface of the PLGA–PBA@MTX nanoparticles may undergo hydrolysis to release MTX for drug delivery application. Indeed, we observed the release of MTX with time in PBS, which is pH-dependent. Thus, before a detailed release study, it became necessary to quantify the drug loading efficiency of PLGA–PBA@MTX for determining the therapeutic efficacy of the nanoparticles. The drug (MTX) loading and conjugation efficiency of PLGA–PBA@MTX were found to be 36.0% and 91.4%, respectively. The MTX conjugation efficiency has been reported by Rostamizadeh et al., who claimed 82% conjugation efficiency of methotrexate within a methoxy poly(ethylene glycol)–poly(caprolactone) copolymer.50 The reason behind the highest MTX loading in the PLGA carrier to the best of our knowledge is the large surface area with EDA-borne functional groups for MTX conjugation. The observed high percentage of MTX loading and conjugation in PLGA–PBA@MTX nanoparticles helped in carrying a sufficient amount of drug for effective therapy even with a lower treatment dose.3.4 Stability of the PLGA–PBA@MTX nanoparticles
The stability of PLGA–PBA@MTX nanoparticles in deionized water is a vital aspect for their long term storage and application in cancer management through their drug release behaviour. It was found that the size of the nanoparticles remained unchanged when they were kept in deionized water as well as pH 7.4 phosphate buffer solution even after 7 days. To study the effect of other pH values on the stability, PLGA–PBA@MTX nanoparticles were suspended in 1× PBS having a wide range of pH values for 7 days. The size of the nanoparticles in pH 4.0, pH 6.0 and pH 9.0 PBS decreased from 252.1 nm to 122 nm, 125.7 nm and 123.7 nm, respectively. The decrease in size of the nanoparticles may be due to either release of MTX or dismantling of the PLGA–PBA@MTX structure. To confirm which mechanism occurred, the sizes of the PLGA–PBA nanoparticles only were measured in this wide range of pH values. It is observed that the size of PLGA–PBA remained almost the same after 7 days in pH 4.0, 6.0 and 9.0 PBS, which confirms that the size of PLGA–PBA@MTX decreased as MTX molecules were dissociated, not because of the dismantling of the PLGA–PBA backbone. Thus, the pH-sensitive amide bonds are responsible for the decrease in size of the PLGA–PBA@MTX nanoparticles at different pH values due to the gradual dissociation of MTX. However, PBA remained undissociated at all these pH values, as covalently conjugated PBA molecules were densely packed into the PLGA matrix after nanoparticle formation. The fluorescence study also confirmed that PBA did not come out from the PLGA matrix as no change in fluorescence in the release medium was observed.3.5 pH-Dependent MTX release
After observing the high loading of MTX, it was worth studying the drug release characteristics of the PLGA–PBA@MTX nanoparticles at pH ∼ 6.5 as this pH prevails in the tumor microenvironment and at ∼4.5, which is found in endosome and lysosome.51,52 To study the release patterns of MTX in the acidic pH range, pH 6.0 and pH 4.0 were chosen to understand the possible mechanism of drug release behaviour in a cellular environment. It was observed that at pH ∼ 6.0, ∼19% MTX was released within 4 h followed by a constant release rate with time till ∼96% MTX was released at 150 h (∼6 days), while at pH 4.0, ∼95% MTX was released within 24 h (Fig. 3d). From the drug release studies, it was realized that both release characteristics, i.e. the initial burst release followed by a sustained release profile, were present at pH ∼ 6.0. In contrast, a fast MTX release occurred at pH 4.0 mainly due to the burst effect, which may be desirable for intratumoral drug delivery. The biphasic release pattern, which was observed at pH 6.0 was similar to the pattern observed by Lu et al.53 They synthesized chitosan (CS)-modified PLGA nanoparticles to overcome the burst release of drug from PLGA nanoparticles. Despite this modification, they observed an initial rapid release of 14.3% and 40.9% paclitaxel after the first 2 h at pH 7.4 and pH 5.5, respectively, and complete release of the drug within 50 h. The 19% burst release of MTX may be due to electrostatically bound MTX molecules on the negatively charged PLGA–PBA surface adsorbed during MTX loading. This observation suggests that apart from amide bonded MTX, there is a possibility of MTX loading on the surface of PLGA–PBA through electrostatic interaction. In the solution phase, the –NH2 functional groups of MTX may remain as –NH3+ that forms electrostatic bonds with the –O–/–OH groups of PLGA–PBA nanoparticles. However, this electrostatic interaction becomes weak as the PLGA–PBA surface gets positively charged at acidic pH, i.e. pH ∼ 6.0, and this introduces a burst release characteristic.22 Unlike CS-modified PLGA nanoparticles, PLGA–PBA@MTX exhibited slower drug release for a longer period of time (∼6 days) to implement the drug-toxicity effect in a sustainable manner. The reason for this behaviour is that the MTX molecules are conjugated with PLGA nanoparticles through amide bonds, and the observed sustained release of MTX at acidic pH may be due to the hydrolysis of the amide bonds present between MTX and the PLGA–PBA nanoparticles through EDA. In general, hydrolysis of the amide bond is difficult at pH ∼ 6.0 to release any molecule. However, as one of the carboxyl groups is in close proximity to the amide function (which undergoes hydrolysis, as shown in Fig. 1), a carboxyl-catalyzed amide bond hydrolysis may occur to facilitate the controlled release of MTX in the cellular compartment like endosome/lysosome where the acidic microenvironment prevailed to increase the therapeutic efficacy of PLGA–PBA@MTX nanoparticles.54The mechanism of MTX release from PLGA–PBA@MTX nanoparticles was analyzed using kinetic modelling. To obtain the best fitting kinetic model, the experimental MTX release data were used in the Korsmeyer–Peppas kinetic model and the release constant parameters like release exponent (n) and regression coefficient (R2) were determined from the best fitting curve (Fig. S7, ESI†). After fitting the MTX release data using the Korsmeyer–Peppas equation, the obtained R2 value was close to 1.0, which indicates that the Korsmeyer–Peppas model is best suited to describe the MTX release. The regression coefficient (R2) and release exponent (n) values are ≈0.99 and ≈0.42, respectively. Since the release exponent value remains <0.5, the Fickian diffusion-based release mechanism for MTX is mainly followed, where the release rate of MTX depends on the rate of hydrolysis of the amide bond and MTX diffusion rate.55
3.6 Hemolytic activity assay
Hemolytic assays were performed to understand the biocompatibility of the PLGA–PBA@MTX nanoparticles in blood, which gives some understanding of in vivo delivery of the nanoparticles to the targeted site. In this study, 0.5 μM, 1.1 μM and 2.0 μM concentrations of PLGA–PBA@MTX were incubated with blood samples and no significant hemolytic activity was observed. The percentage hemolysis was well within the 5% range (Fig. S8, ESI†). These results suggested that PLGA–PBA@MTX could be used as a carrier for delivery of chemotherapeutic agents to the cancer site, while evidencing its own low toxicity toward non-cancerous cells.3.7 PLGA–PBA@MTX nanoparticle-induced cytotoxicity studies
The PLGA–PBA@MTX nanoparticles were designed to increase the overall delivery efficacy of MTX in reduced folate carrier (RFC) lacking cells. Normally, MTX is delivered into cells predominantly via the RFC, which is a bi-directional anion exchanger with 12 putative transmembrane domains. In the cellular environment, MTX inhibits the synthesis of nucleotides by binding irreversibly with the enzymes, i.e. dihydrofolate reductase (DHFR), thymidylate synthase (TS), glycinamide ribonucleotide transformylase (GAR transformylase) and 5-aminoimidazole-4-carboxamide ribonucleotide transformylase (AICAR transformylase), essential for the synthesis of thymidylate (dTMP), adenylate (AMP) and guanylate (GMP), respectively, which eventually disrupts the DNA synthesis followed by apoptosis-mediated cell death. Additionally, MTX hinders the synthesis of methionine and S-adenosyl-methionine required for the methylation of DNA, histones and lipids.17,56 In this work, we have considered MDA-MB-231 and MCF-7, which are MTX-resistant breast cancer cell lines. The reason for MTX resistance in MDA-MB-231 cells is mostly the impaired drug transport phenomenon as the cells do not express RFC mRNA.57 On the other hand, MCF-7 cells become resistant to MTX due to the increased levels of DHFR in cytosol.58 To assess whether PLGA–PBA@MTX nanoparticles have any cytotoxic effect, we determined the cell viability of MDA-MB-231 and MCF-7 cells in the presence of free MTX and PLGA–PBA@MTX nanoparticles. For this purpose, each cell line was treated with MTX and PLGA–PBA@MTX nanoparticles with conjugated MTX concentrations ranging from 15 nM to 2.6 μM for MDA-MB-231 and from 5 μM to 80 μM for MCF-7. The concentration of MTX in PLGA–PBA@MTX was calculated from the calibration curve (absorbance versus MTX concentration), as shown in the ESI.† The cell viability data are represented as mean ± SD, obtained from three independent experiments (Fig. S9, ESI†). Within the applied drug concentration range, MTX exhibits minimum cytotoxicity in MDA-MB-231 and MCF-7 cells, which is also supported by other previously published reports.58 However, when treated with PLGA–PBA@MTX nanoparticles, a drastic reduction of cell viability was observed with IC50 values of ∼1.1 μM for MDA-MB-231 (Fig. S9a, ESI†) and ∼16 μM for MCF-7 (Fig. S9b, ESI†) after 48 h of treatment. The study indicates that MTX did not exhibit significant cytotoxic properties at lower concentrations, but when conjugated with PLGA–PBA@MTX nanoparticles, the cytotoxic behaviour increases significantly even at low concentration. We assumed that a large number of MTX molecules were hydrolyzed from the surface of the PLGA–PBA@MTX nanoparticles after entering the cells, due to the acidic environment of the endosome/lysosome, and inhibited the synthesis of the nucleotides by binding irreversibly with the required enzymes, leading to significant DNA damage and cell death.3.8 Cellular internalization pathway of PLGA–PBA@MTX nanoparticles
There are several transport mechanisms, such as passive diffusion, active transport or endocytosis, that can be used to internalize small molecules and nanoparticles inside cells. Endocytosis is a temperature-sensitive process, which is mainly of two types: phagocytosis and pinocytosis.59 Phagocytosis is primarily observed in macrophages, while pinocytosis is found in all other cell types. Pinocytosis may be classified into four categories, clathrin-dependent endocytosis, macropinocytosis, caveolae-dependent endocytosis, and clathrin/caveolae-independent endocytosis.59 PLGA–PBA@MTX nanoparticles should utilize one or more of these pathways for cellular internalization, which was monitored by the PBA derived fluorescence property of the nanoparticles.PLGA–PBA@MTX was designed to deliver the drug inside cells that are drug resistant due to a lack of RFC expression. To investigate whether endocytosis is involved in the intracellular transport of PLGA–PBA@MTX, we measured the fluorescence of the nanoparticle-treated MDA-MB-231 and MCF-7 cells at 37 °C (control cells) and 4 °C for 2 h (Fig. 4ai and ii, bi and ii). Negligible fluorescence was observed in nanoparticle-treated cells incubated at 4 °C (Fig. 4aii and bii), while a significant increase in fluorescence intensity was observed at 37 °C (Fig. 4ai and bi). The quantification of fluorescence intensities from the reproducible experiments indicated that PLGA–PBA@MTX nanoparticles exhibit significantly high internalization after 2 h of incubation (*p < 0.0001) at 37 °C in comparison to the cells incubated at 4 °C, suggesting the involvement of endocytosis in the intracellular uptake of PLGA–PBA@MTX nanoparticles in both MDA-MB-231 and MCF-7 cells. After confirming the involvement of endocytic pathways; we blocked different endocytic processes with chemical inhibitors to understand the mechanism of endocytosis. For this purpose, MDA-MB-231 and MCF-7 cells (pre-exposed to different inhibitors with concentrations that are non-toxic or have very low toxicity to the cells) were incubated with PLGA–PBA@MTX for 2 h and the fluorescence intensities were monitored. It was observed that both MDA-MB-231 and MCF-7 cells pre-incubated with chlorpromazine show a similar uptake of nanoparticles to the control cells (cells treated at 37 °C) as they did not show any significant difference in fluorescence intensity (Fig. 4aiii and biii). Thus, the involvement of any clathrin raft-mediated endocytosis was eliminated in this study. However, when both cells were treated with lovastatin, which blocks caveolae-dependent endocytosis, MDA-MB-231 cells showed less fluorescence intensity compared to the control cells (Fig. 4aiv), which suggests the partial involvement of lipid raft-mediated endocytosis in the transport of PLGA–PBA@MTX nanoparticles into MDA-MB-231 cells, but MCF-7 did not show any significant decrease in fluorescence intensity compared to the control cells (Fig. 4biv). This observation corroborates the results of some previous studies, which showed a higher expression of FR-α in MDA-MB-231 cells than in MCF-7 cells and involvement of the non-classical lipid raft-mediated endocytosis pathway in FR-α-mediated internalization of folate, which does not involve clathrin-coated pits.16,60
 | ||
| Fig. 4 Internalization pathway of PLGA–PBA@MTX nanoparticles in (a) MDA-MB-231 cells and (b) MCF-7 cells analyzed by fluorescence microscopy under different conditions i.e. (i) 37 °C and (ii) 4 °C and after (iii) chlorpromazine, (iv) lovastatin and (v) amiloride treatment. Cells were visualized under bright field and DAPI channels. (c) Quantification of fluorescence intensity of PLGA–PBA@MTX in MDA-MB-231 cells depicting a significant decrease in internalization in amiloride- and lovastatin-treated cells, compared with the control group; and chlorpromazine-treated cells showed no significant change in fluorescence intensities. (d) Quantification of fluorescence intensity of PLGA–PBA@MTX in MCF-7 showing a significant decrease in internalization in amiloride-treated cells, compared with the control group, whereas chlorpromazine- and lovastatin-treated cells showed no significant change in fluorescence intensities. | ||
Although we have observed significant internalization by FR-α-dependent lipid raft-mediated endocytosis in MDA-MB-231 cells, it is unlikely to cause apoptosis singlehandedly, as demonstrated in the MTT assay (Fig. S9a, ESI†) in the given concentration range. Thus, the involvement of a second mode of internalization was anticipated. As expected, pre-incubation with amiloride completely blocked the uptake of PLGA–PBA@MTX nanoparticles in MDA-MB-231 and MCF-7 cells compared to the control cells (Fig. 4av and bv). These results established that the intracellular uptake of the nanoparticles in MDA-MB-231 cells by both macropinocytosis and lipid raft-mediated endocytosis pathways triggered cell death. In MCF-7 cells, intracellular uptake of the nanoparticles occurred mainly by macropinocytosis, which may be due to the low level of expression of FR-α on the cell surface.
3.9 Endosome staining with acridine orange to demonstrate the co-localization of PLGA–PBA@MTX nanoparticles in endosomes
The internalization study showed that the transport of PLGA–PBA@MTX nanoparticles occurred mainly through macropinocytosis in both cells, i.e. MDA-MB-231 and MCF-7, and by FRα-mediated endocytosis into MDA-MB-231 cells. It is reported that if endocytosis occurs, then nanoparticles will remain in endosome and lysosome.61 Romero et al. reported the internalization of PLGA nanoparticles via macropinocytosis and their co-localization with endosome.62 Because of these results, we also anticipated that PLGA–PBA@MTX would remain in endosomes. To confirm the localization of the nanoparticles in endosome like cellular compartments after endocytosis, acridine orange staining was performed to visualize the endosomes and lysosomes formed after endocytosis.63 Acridine orange specifically stains endosomes with red fluorescence and nuclei with green fluorescence. The co-localization of PLGA–PBA@MTX nanoparticles was evaluated to determine their presence in endosomes using the emission maximum of the nanoparticles at 378 nm, which is the wavelength of the blue fluorescence region. In the case of the control cells (untreated), blue fluorescence dots were absent due to the lack of PLGA–PBA@MTX nanoparticles, and the merged image showed no fluorescence color deviation from red (Fig. 5). However, in the case of PLGA–PBA@MTX-treated cells, blue fluorescence dots were well-observed for the nanoparticles and the merged image showed a deviation of color from blue and red to purple (Fig. 5). This shift in color in the merged image confirmed the co-localization of PLGA–PBA@MTX nanoparticles and endosome, which occurred due to the internalization through micropinocytosis and caveole-mediated endocytic pathways. | ||
| Fig. 5 Co-localization of the internalized PLGA–PBA@MTX nanoparticles in endosomes of MDA-MB-231; merged image of PLGA–PBA@MTX-treated cells showing deviation of color from blue (due to nanoparticle fluorescence) and red (due to endosome staining with acridine orange) to purple, while the merged image of the control cells shows no deviation of color. | ||
3.10 Mode of PLGA–PBA@MTX nanoparticle-induced cell death
The previous cellular studies clearly depicted internalization of the PLGA–PBA@MTX nanoparticles in both RFC positive (MCF-7) and negative (MDA-MB-231) cells. In order to assess the therapeutic effect of PLGA–PBA@MTX nanoparticles after internalization, the apoptosis assay was performed using an Annexin V Alexa Fluor 488/PI staining process and detected through flow cytometry. Annexin V Alexa Fluor 488 was used to stain the apoptosis-specific flipped membrane phospholipids, i.e. phosphatidylserine (PS), which are usually present on the cytoplasmic side of the plasma membrane, while in apoptotic cells, they are exposed on the outer surface due to membrane flipping. The cell membrane permeability of necrotic cells was stained using PI. In this study, MDA-MB-231 and MCF-7 cells were treated with PLGA–PBA@MTX and then stained with Annexin –V Alexa Fluor 488/PI after 48 h of treatment. The fluorescence intensity of Annexin V Alexa Fluor 488 was increased in both cases of MDA-MB-231 and MCF-7 cells after treatment with the nanoparticles in a dose-dependent manner. Three different conjugated MTX concentrations of the nanoparticles were tested and found to cause significantly high apoptosis (*p < 0.0001) compared with the control group (untreated) (Fig. S10a–e in MDA-MB-231 and Fig. S11a–e in MCF-7, ESI†). These results suggested that the present nanoformulation strategy can be an effective route in the treatment of RFC-deficient MTX resistant cancer as well as other metastatic breast cancers.3.11 PLGA–PBA@MTX nanoparticle-induced DNA damage
As we observed that MTX is released from the nanoparticle surface through hydrolysis of the amide bond, there would be a possibility that a similar release behavior could occur inside the acidic cellular compartments and in cytoplasm. In the previous literature, it was reported that MTX irreversibly binds with the essential enzymes of nucleotide biosynthesis like dihydrofolate reductase, thymidylate synthase, GAR transformylase and AICAR transformylase with a high affinity, which eventually inhibit the synthesis of thymidylate, adenylate and guanylate, respectively, necessary for nucleotide synthesis.17,56 In the absence of these precursor nucleotides, DNA synthesis is disrupted, leading to DNA damage.23 In this study, if MTX release occurs from PLGA–PBA@MTX in the cell cytoplasm, we would then observe damage of DNA due to the MTX-mediated inhibition of nucleotide synthesis. To validate this hypothesis, we have performed comet assay to demonstrate the DNA damage induced by PLGA–PBA@MTX nanoparticles. In the electrophoresis-based comet assay, the “head” part indicates the undamaged DNA nucleoid and the “tail” part signifies the damaged DNA strip trailed behind the “head”.64 The percentage of DNA in the tail is directly proportional to the percentage of DNA damage that has occurred in a particular cell. MDA-MB-231 cells treated with PLGA–PBA@MTX nanoparticles showed a significantly high amount of DNA in the tail part compared to the control and MTX-treated cells (Fig. 6). This justifies that PLGA–PBA@MTX nanoparticles have the ability to induce DNA damage and, consequently, cell-killing in a much more efficient way than MTX at a given concentration.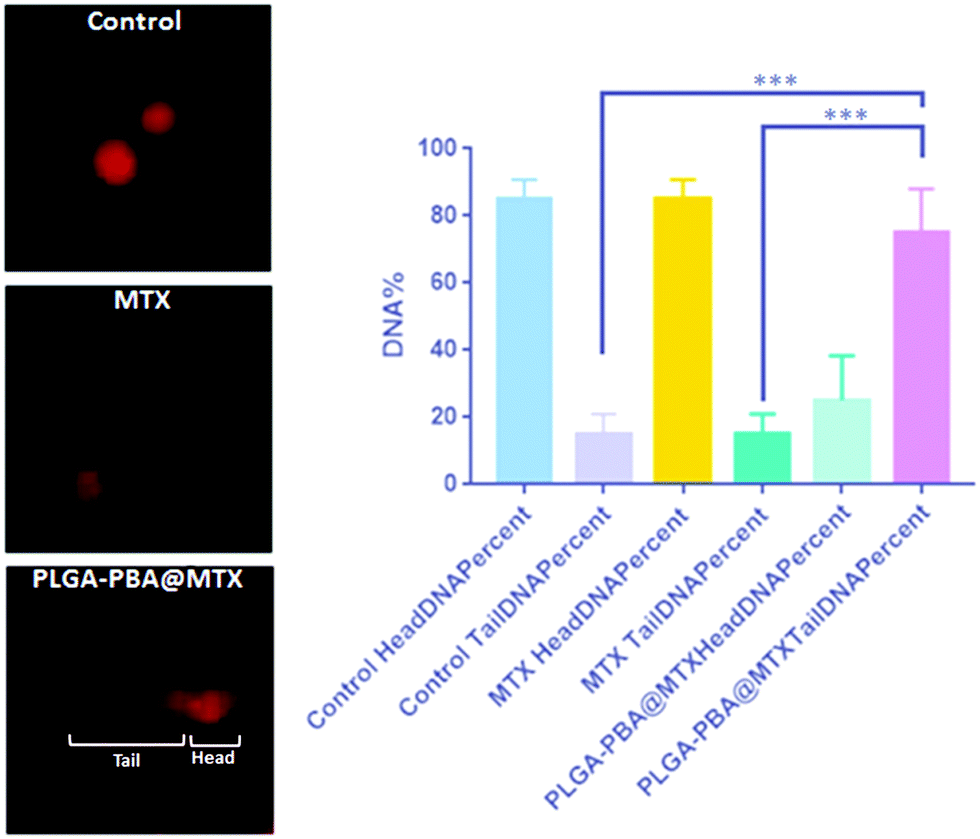 | ||
| Fig. 6 Electrophoresis-based comet assay to demonstrate the DNA damage induced by PLGA–PBA@MTX nanoparticles. The image shows the comet formation including the head (undamaged DNA) and tail (damaged DNA) with the statistical analysis of their percentages. | ||
4. Conclusions
The present study demonstrates a novel approach to the synthesis of PLGA-based nanoparticles with fluorescence and therapeutic properties to treat drug-resistant breast cancer cells. The stable fluorescence property was developed by the covalent conjugation of PBA molecules with PLGA polymer chains via EDA molecules as a bridge between them. The covalent conjugation of PBA through amide bonding in the PLGA–PBA polymer was established by FTIR, NMR, and mass spectroscopic analyses. When PLGA–PBA polymer was electrosprayed at 8.0 kV, fluorescent nanoparticles were obtained with mono-dispersity (40 nm in size), a spherical shape, and a smooth texture of the surface, as characterized by AFM, FESEM and TEM observation. To make drug conjugated PLGA–PBA nanoparticles, the carboxylic group of the MTX molecule was amide-bonded with –NH2 groups of the EDA-conjugated PLGA–PBA nanoparticles. As MTX molecules conjugated on the surface of the PLGA–PBA nanoparticles, the size of the PLGA–PBA@MTX nanoparticles became ∼105 nm with 36% drug loading and 91.4% conjugation efficiency. The high conjugation of MTX and the outward protrusion of the free –NH2 groups of the attached MTX molecules helped in multivalent interaction with FAα receptors, followed by internalization inside cells. This effectiveness of PLGA–PBA@MTX nanoparticles was reflected in the cytotoxicity study and also in the apoptosis assay. The IC50 values of the PLGA–PBA@MTX nanoparticles were 1.1 μM and 16.0 μM in MDA-MB-231 and MCF-7 cells, respectively, which were considerably lower than the IC50 value of free MTX. The MTX release profile from PLGA–PBA@MTX nanoparticles was studied in the release medium of acidic pH like the pH observed in the tumor microenvironment and endosome/lysosome. The release of MTX from PLGA–PBA@MTX nanoparticles at pH 6.0 followed the Korsmeyer–Peppas kinetics model with a Fickian diffusion mechanism. At pH below 6, which is found in the endosome, ∼95% MTX release was observed within 24 h, which was also in accordance with the MTT and apoptosis assay results. Significant internalization of PLGA–PBA@MTX nanoparticles via the macropinocytosis pathway in MTX-resistant MDA-MB-231 and MCF-7 cells was established by monitoring the fluorescence of PBA in the nanoparticles. In MDA-MB-231 cells, the internalization of PLGA–PBA@MTX nanoparticles also followed the lipid raft-mediated endocytosis pathway through FR-α receptors to induce higher cell cytotoxicity than that in MCF-7 cells. All these results concluded that PLGA–PBA@MTX nanoparticles are highly efficient in delivering MTX and also executing apoptosis-mediated cell death of drug-resistant breast cancer cells. Thus, there is enormous future potential of this novel PLGA-based nanoparticle, which can be used for the loading of MTX-type drug molecules in a nanoparticle matrix and conjugation of target-specific small molecules or antibodies on the surface of the nanoparticle system for theranostic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are thankful to the Head, Materials Processing and Microsystems Lab, and the Director, CSIR-CMERI, Durgapur, for the support and encouragement. Support from CSIR-SRF (CSIR-HRDG) sanction letter no. 31/19(0010)/2018-EMR-I is gratefully acknowledged. Dr Biswarup Basu gratefully acknowledges The Director, CNCI, for infrastructural support and would like to thank the Science and Engineering Research Board, Govt. of India, for partial funding (SB/YS/LS-171/2014) to carry out the cell studies.References
- F. Bray, J. Ferlay, I. Soerjomataram, R. Siegel, L. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 Search PubMed.
- R. Wang, Y. Zhu, X. Liu, X. Liao, J. He and L. Niu, BMC Cancer, 2019, 19, 1–12 Search PubMed.
- J. T. Jørgensen, Ann. Transl. Med., 2019, 7, 148 CrossRef.
- J. L. Meisel, V. A. Venur, M. Gnant and L. Carey, ASCO Educ. B, 2018, 38, 78–86 Search PubMed.
- S. Senapati, A. K. Mahanta, S. Kumar and P. Maiti, Signal Transduction Targeted Ther., 2018, 3, 1–19 CrossRef CAS.
- B. Fortuni, T. Inose, M. Ricci, Y. Fujita, I. Van Zundert, A. Masuhara, E. Fron, H. Mizuno, L. Latterini, S. Rocha and H. Uji-i, Sci. Rep., 2019, 9, 1–13 CrossRef CAS.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS.
- A. Thomas, T. Appidi, A. B. Jogdand, S. Ghar, K. Subramaniyam, G. Prabusankar, J. R. Mohanty and A. K. Rengan, ACS Appl. Polym. Mater., 2020, 2, 1388–1397 CrossRef CAS.
- E. Calzoni, A. Cesaretti, A. Polchi, A. Di Michele, B. Tancini and C. Emiliani, J. Funct. Biomater., 2019, 10, 1–15 Search PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Préat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS.
- H. S. Yoo and T. G. Park, J. Controlled Release, 2001, 70, 63–70 CrossRef CAS.
- X. Liu, A. L. Miller, M. J. Yaszemski and L. Lu, RSC Adv., 2015, 5, 33275–33282 RSC.
- C. W. Liu and W. J. Lin, Int. J. Nanomed., 2012, 7, 4749–4767 CAS.
- L. Wang, G. Bian, L. Wang, L. Dong, H. Chen and T. Xia, Spectrochim. Acta, Part A, 2005, 61, 1201–1205 CrossRef.
- N. H. Ly, T. D. Nguyen, T. L. Bui, S. Lee, J. Choo and S. W. Joo, Colloids Surf., A, 2017, 518, 295–303 CrossRef.
- A. Cheung, H. J. Bax, D. H. Josephs, K. M. Ilieva, G. Pellizzari, J. Opzoomer, J. Bloomfield, M. Fittall, A. Grigoriadis, M. Figini, S. Canevari, J. F. Spicer, A. N. Tutt and S. N. Karagiannis, OncoTargets Ther., 2016, 7, 52553–52574 CrossRef.
- P. Koźmiński, P. K. Halik and E. Gniazdowska, Encyclopedia, 2020, 1–5 Search PubMed.
- G. Turashvili and E. Brogi, Front. Med., 2017, 4, 227 CrossRef.
- J. Worm, A. F. Kirkin, K. N. Dzhandzhugazyan and P. Guldberg, J. Biol. Chem., 2001, 276, 39990–40000 CrossRef CAS.
- P. T. Wong and S. K. Choi, Int. J. Mol. Sci., 2015, 16, 1772–1790 CrossRef CAS.
- E. Nogueira, M. P. Sárria, N. G. Azoia, E. Antunes, A. Loureiro, D. Guimarães, J. Noro, A. Rollett, G. Guebitz and A. Cavaco-Paulo, Biochemistry, 2018, 57, 6780–6786 CAS.
- R. Vivek, R. Thangam, V. Nipunbabu, C. Rejeeth, S. Sivasubramanian, P. Gunasekaran, K. Muthuchelian and S. Kannan, ACS Appl. Mater. Interfaces, 2014, 6, 6469–6480 CAS.
- A. H. Borchers, K. A. Kennedy and J. A. Straw, Cancer Res., 1990, 50, 1786–1789 CAS.
- Y. K. Hye Sung, H. O. Ham, Y. J. Son, P. Messersmith and H. Sang, J. Mater. Chem. B, 2013, 1, 3940–3949 RSC.
- M. Chatterjee, N. Jaiswal, A. Hens, N. Mahata and N. Chanda, Mater. Sci. Eng., C, 2020, 114, 111029 CrossRef CAS.
- C. K. Dixit, S. K. Vashist, B. D. Maccraith and R. O. Kennedy, Nat. Protoc., 2011, 6, 439–445 CrossRef CAS.
- A. V. Aseichev, O. A. Azizova, E. M. Beckman, O. I. Skotnikova, L. B. Dudnik, O. N. Shcheglovitova and V. I. Sergienko, Bull. Exp. Biol. Med., 2014, 156, 495–498 CrossRef CAS.
- R. Maity, M. Chatterjee, A. Banerjee, A. Das, R. Mishra, S. Mazumder and N. Chanda, Mater. Sci. Eng., C, 2019, 104, 109909 CrossRef CAS.
- V. Maréchal, M.-C. Prevost, C. Petit, E. Perret, J.-M. Heard and O. Schwartz, J. Virol., 2001, 75, 11166–11177 CrossRef.
- E. J. Smart and R. G. W. Anderson, Methods Enzymol., 2002, 353, 131–139 CAS.
- L. H. Wang, K. G. Rothberg and R. G. Anderson, J. Cell Biol., 1993, 123, 1107–1117 CrossRef CAS.
- D. Suresh, A. Zambre, N. Chanda, T. J. Hoffman, C. J. Smith, J. D. Robertson and R. Kannan, Bioconjugate Chem., 2014, 25, 1565–1579 CrossRef CAS.
- R. Matteoni and T. E. Kreis, J. Cell Biol., 1987, 105, 1253–1265 CrossRef CAS.
- S. Nandhakumar, S. Parasuraman, M. Shanmugam, K. Rao, P. Chand and B. Bhat, J. Pharmacol. Pharmacother., 2011, 2, 107 CrossRef CAS.
- J. Wang, H. B. Liu, S. Park, S. Y. Kim, T. Joo and C. S. Ha, RSC Adv., 2012, 2, 4242–4249 RSC.
- N. Jaiswal, A. Hens, M. Chatterjee, N. Mahata and N. Chanda, J. Colloid Interface Sci., 2019, 534, 122–130 CrossRef CAS.
- T. I. Croll, A. J. O’Connor, G. W. Stevens and J. J. Cooper-White, Biomacromolecules, 2004, 5, 463–473 CrossRef CAS.
- M. Chatterjee, A. Hens, K. Mahato, N. Jaiswal, N. Mahato, Nagahanumaiah and N. Chanda, J. Colloid Interface Sci., 2017, 506, 126–134 CrossRef CAS.
- S. Philip and S. Kuriakose, J. Fluoresc., 2019, 29, 387–398 CrossRef CAS.
- E. D. Pereira, R. Cerruti, E. Fernandes, L. Peña, V. Saez, J. C. Pinto, J. A. Ramón, G. E. Oliveira and F. G. De Souza, Polimeros, 2016, 26, 137–143 CrossRef.
- S. Majeed, V. Filiz, S. Shishatskiy, J. Wind, C. Abetz and V. Abetz, Nanoscale Res. Lett., 2012, 7, 296 CrossRef.
- H. Xiao and L. Wang, Int. J. Nanomed., 2015, 10, 5309–5325 CrossRef CAS.
- S. Jain, V. V. Rathi, A. K. Jain, M. Das and C. Godugu, Nanomedicine, 2012, 7, 1311–1337 CrossRef CAS.
- N. Arsalani, P. Nezhad-Mokhtari and E. Jabbari, Artif. Cells, Nanomed. Biotechnol., 2019, 47, 540–547 CrossRef CAS.
- L. Wang, X. Xie, D. Liu, X. Bin Fang, P. Li, J. B. Wan, C. W. He and M. W. Chen, RSC Adv., 2016, 6, 28331–28342 RSC.
- Y. Yan, J. Cui, S. Zhao, J. Zhang, J. Liu and J. Cheng, J. Mater. Chem., 2012, 22, 1928–1936 RSC.
- Y. Zhang, T. P. Thomas, A. Desai, H. Zong, P. R. Leroueil, I. J. Majoros and J. R. Baker, Bioconjugate Chem., 2010, 21, 489–495 CrossRef CAS.
- S. F. Peng, C. Y. Lee, M. J. Hour, S. C. Tsai, D. H. Kuo, F. A. Chen, P. C. Shieh and J. S. Yang, Int. J. Oncol., 2014, 44, 238–246 CrossRef CAS.
- P. Guo, X. Wang, L. Liu, M. G. Belinsky, G. D. Kruh and J. M. Gallo, J. Pharm. Biomed. Anal., 2007, 43, 1789–1795 CrossRef CAS.
- K. Rostamizadeh, M. Manafi, H. Nosrati, H. Kheiri Manjili and H. Danafar, New J. Chem., 2018, 42, 5937–5945 RSC.
- S. Uthaman, K. M. Huh and I. K. Park, Biomater. Res., 2018, 22, 1–11 CrossRef.
- J. Liu, Y. Huang, A. Kumar, A. Tan, S. Jin, A. Mozhi and X. J. Liang, Biotechnol. Adv., 2014, 32, 693–710 CrossRef CAS.
- B. Lu, X. Lv and Y. Le, Polymers, 2019, 11, 304 CrossRef.
- I. V. Komarov, A. Y. Ishchenko, A. Hovtvianitsa, V. Stepanenko, S. Kharchenko, A. D. Bond and A. J. Kirby, Molecules, 2019, 24, 572 CrossRef.
- M. Dhanka, C. Shetty and R. Srivastava, Int. J. Biol. Macromol., 2017, 110, 346–356 CrossRef.
- J. Wohlrab, R. H. H. Neubert, J. Michael and S. Naumann, JDDG - J. Ger. Soc. Dermatology, 2015, 13, 891–902 Search PubMed.
- R. Yang, W. W. Li, B. H. Hoang, H. Kim, D. Banerjee, A. Kheradpour, J. H. Healey, P. A. Meyers, J. R. Bertino and R. Gorlick, BMC Cancer, 2008, 8, 124 CrossRef.
- E. Selga, C. Oleaga, S. Ramírez, M. C. de Almagro, V. Noé and C. J. Ciudad, Genome Med., 2009, 1, 83 CrossRef.
- T.-G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS.
- J. P. Marshalek, P. S. Sheeran, P. Ingram, P. A. Dayton, R. S. Witte and T. O. Matsunaga, J. Controlled Release, 2016, 243, 69–77 CrossRef CAS.
- J. P. Lim and P. A. Gleeson, Immunol. Cell Biol., 2011, 89, 836–843 CrossRef CAS.
- G. Romero, D. J. Sanz, Y. Qiu, D. Yu, Z. Mao, C. Gao and S. E. Moya, J. Mater. Chem. B, 2013, 1, 2252–2259 RSC.
- A. Evangelatov, R. Skrobanska, N. Mladenov, M. Petkova, G. Yordanov and R. Pankov, Drug Delivery, 2014, 23, 1–10 Search PubMed.
- S. R. Beedanagari, S. V. Vulimiri, S. P. Bhatia and B. Mahadevan, Biomarkers in Toxicology, Elsevier Inc., 2014, pp. 729–742 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Hydrodynamic size, zeta potential (ζ), fluorescence quenching study of PBA by MTX, FTIR spectra, 1H NMR spectra, mass spectroscopy drug release kinetics, conjugation efficiency calculation, MTT assay and results of co-localization in endosomes supplied. See DOI: 10.1039/d0ma00646g |
| This journal is © The Royal Society of Chemistry 2020 |