
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High-throughput fabrication of hepatic cell clusteroids with enhanced growth and functionality for tissue engineering applications†
Anheng
Wang
 a,
Leigh A.
Madden
b and
Vesselin N.
Paunov
*a
a,
Leigh A.
Madden
b and
Vesselin N.
Paunov
*a
aDepartment of Chemistry and Biochemistry, University of Hull, Hull, HU67RX, UK. E-mail: v.n.paunov@hull.ac.uk; Tel: +44 1482 465660
bDepartment of Biomedical Sciences, University of Hull, Hull, HU67RX, UK
First published on 6th November 2020
Abstract
Culturing of cells as three-dimensional (3D) clusters can enhance in vitro tests for basic biological research as well as for therapeutics development. Such 3D culture models, however, are often more complicated, cumbersome and expensive than two-dimensional (2D) cultures. Current methods for the preparation of tissue spheroids require complex materials, involve tedious facilities and are generally not scalable. We report a novel inexpensive and up-scalable method for the preparation of large quantities of viable cell clusters (clusteroids) of hepatocytes (Hep-G2). The method has a high throughput potential and is based on an aqueous two-phase system (ATPS) of stable water-in-water (w/w) Pickering emulsions, formed of dextran (DEX) drops and poly ethylene oxide (PEO) continuous phase stabilized with whey protein particles. This system enabled the rapid fabrication of cell clusteroids from Hep-G2 cells. Here, the interfacial tension of the aqueous phase in the emulsion droplets, where the cells partition preferentially, is used to wrap the cells in separate compartments, and then the droplets are shrank by changing the balance of ATPS, thus rapidly driving the cells from larger and loosely packed DEX drops to mostly spherical clusters. Cell–Cell adhesion was strongly promoted within the w/w Pickering emulsion droplets which helped the formation of the 3D clusteroids. These were collected after subsequent dilution of the emulsion with culture media. The collected hepatic clusteroids were incorporated into an alginate hydrogel in media to study their proliferation and the cell function compared with individual cells under the same conditions. Our results confirmed that urea and albumin production, which are both linked to hepatocyte cell function, was strongly reinforced in the clusteroid based tissues compared to the one formulated with individual cells. This methodology could potentially extend the w/w emulsion cell clustering platform in tissue generation and preparation of large quantities of organoids for drug tests and replacement of animal models.
1. Introduction
The liver is the major organ involved in the metabolism and clearance of numerous xenobiotics. However, detoxification of many of these xenobiotics can contribute to the impairment of liver functionality resulting in injury.1 Understanding xenobiotic safety is important to numerous industries such as pharmaceuticals, agrochemicals, industrial chemicals, and consumer products, and therefore robust systems for xenobiotic assessment are required, in particular, to support the early research phases of product development.2 Adverse drug reactions (ADRs) represent a considerable hindrance to the amelioration of novel therapeutics with ∼21% of drug attrition attributed to toxicity during the development process.3 ADRs are a major cause of liver injury and are responsible for up to 7% of hospital admissions. Drug-induced liver injury (DILI), one of the most common forms of ADRs, is the main reason for the withdrawal of drugs from the market.3–5 These reactions are complicated and often require interactions between the multiplex of parenchymal hepatocytes and non-parenchymal cells such as stellate cells (SCs), Kupffer cells (KCs), liver sinusoidal endothelial cells (LSECs), etc. DILI encompasses a vast spectrum of manifestations: the impairment of mitochondrial function, inflammation and lethal effects of the immune response, cell death via necrosis and apoptosis, and pathologies including micro-vesicular steatosis and cholestasis.6In vitro liver models that possess the capability to predict potential adverse liver manifestations are greatly valued in the pharmaceutical sector7 as well as other industries.To expand the supply of available livers for transplant, numerous surgical options have been pursued, including split liver transplants and living-related partial donor procedures.8 In spite of these surgical advances and improvements in organ allocation, organ shortages remain acute, suggesting that it is unlikely that liver transplantation procedures alone will ever meet the increasing demand. Therefore, cell-based therapies have long-held promise as an alternative to organ transplantation. Biomedical researchers at present have several types of 3D cell models at their disposal. One such model most resembling native tissues is organotypic explants, a small piece of tissue freshly dissected from experimental animals and maintained in vitro.9 Tissue explants have been exploited in nearly all biomedical research fields and probably will not be replaced by other 3D culture models in the near future.
However, such a model faces major obstacles, such as difficulty in obtaining specimens and ethical approval. In addition, tissue explants are typically several millimetres in size, which hampers nutrient transport to central regions and results in poor tissue viability. In view of these problems, a 3D culture model that is simple to prepare and on a scale of 100–500 μm would be a useful practical alternative to tissue explants or drug testing platforms. In order to control the size of individual spheroids, hanging-drop culture10,11 and 3D culturing in microwells12 are extensively used. Alternative techniques are based on micro-rotational flow13 and the magneto-Archimedes effect14 to form similarly sized tissue spheroids. The current methods for the production of cellosomes15,16 could also potentially serve as a possible route for the large scale production of desired cell spheroids. Encapsulation of cells in such structures has also been achieved using natural biopolymers like alginate, pectin, agarose and chitosan.17 However, most of the current processes for the generation of cell spheroids cannot be practically used for tissue engineering as they are required to be produced on a large scale. A new cheap and rapid technology is needed, capable of high throughput spheroid production.
Aqueous two-phase systems (ATPS) have previously been used to make tumouroids,12,18,19 mainly by distributing dextran-rich drops (DEX) and tumour cells to a polyethylene glycol rich aqueous medium (PEG). Isolated tumour cells have been cultured into individual globules in these DEX drops and their response to drug therapy has been further studied12,20–22 since higher chemotherapeutic drug resistance has been observed when globules manufactured using this technique are compared to 2D cultured globules.19 Previous techniques have produced effective tumour spheroids to study drug efficiency, however, the aim of these studies is to create large spheroids that should essentially have a necrotic core and cells proliferating on the surface of the spheroid. However, in tissue engineering (TE) applications, there are technical limits on the scalability of these methods in the production of a large number of cell spheroids. Recently, Das et al. reported a rapid, expandable technique for the production of tissue globules from human embryonic kidney cells (HEK293) by trapping them in w/w Pickering emulsion droplets.23,24 Their technique is based on the formation of w/w Pickering emulsions, in which cells are efficiently distributed in emulsion droplets. In their method, the interfacial tension of the aqueous DEX phase in the droplets, where the cells partition preferentially, is used to wrap the cells in separate compartments, and then the droplets are shrank by changing the osmotic balance of the two aqueous phases (ATP), thus rapidly driving the cells from larger and loosely packed drops to mostly spherical cell clusters. These are collected after the break down of the w/w emulsion by subsequent dilution with a growth media.
Intending to generate cell clusters with enhanced function, we developed further the method of Das et al.23 and Celik et al.24 for the preparation of hepatocyte cell clusters (clusteroids) using w/w Pickering emulsions as templates and explored their formulation in suitable hydrogel matrix for 3D tissue engineering of the liver graft. 3D cell cultures of human adult hepatocyte cells (Hep-G2) were used for the production of hepatocyte spheroids. This methodology could be used as a blueprint to enable the growth of patients’ liver cells and to fabricate autologous cell clusteroids for potential clinical applications. Herein, the present work was conducted to discuss the materials and techniques used to control the size of the emulsion drop templates and the average size of the resulting hepatocyte clusteroids. Our results show that viable hepatocyte clusteroids can be easily produced using this technique in large quantities. We demonstrate that the clusteroids fabricated by our w/w Pickering emulsion templating enable the hepatic cells to gain higher growth rate, which also results in clearly enhanced albumin and urea production.
2. Materials and methods
2.1 Materials
Deionized water purified by reverse osmosis and ion exchange using a Milli-Q water system (Millipore) was used in all our studies. Dextran (MW 500 kDa) and PEO (MW 200 kDa) were both purchased from Sigma-Aldrich. Whey protein was sourced from No1. Supplements, Suffolk, UK. Fluorescein diacetate (FDA, 98%) and sodium alginate and Corning® Transwell® polyester membrane cell culture inserts (12 mm, 12 well plates) were purchased from Sigma-Aldrich, UK. Sodium chloride (99.8%) and calcium chloride were purchased from Fisher Scientific. Eagle's Modified Eagle Medium (EMEM) and Trypsin–EDTA were sourced from Gibco®, Fisher Scientific. NUNC Cell culture 24-well plates were purchased from Thermo Fisher Scientific. Urea and albumin ELISA kit were purchased from Sigma-Aldrich. Fetal bovine serum (FBS, Labtech, Heathfield, UK) 0.25% Trypsin–EDTA (1X, Lonza). Alginate lyase was purchased from Sigma Aldrich. All other chemicals were of analytical grade.2.2 2D Hep-G2 cell culture
Hep-G2 cell line culture was purchased from Public Health England (PHE, Culture collection 85011430 Hep G2) cultured from growing cells. The cells were cultured in EMEM media supplemented with 10% FBS and placed in an incubator (37 °C, 5% CO2). After reaching 80% confluence, Hep-G2 cells were carefully washed with phosphate buffer saline (PBS, Labtech, UK) for 10 s and then incubated with 0.25% Trypsin–EDTA (1X, Lonza, UK), which allowed us to detach the cells from their support after 5 min. Its action was neutralized by adding complete EMEM medium before centrifugation at 400 × g for 4 min. After resuspension in fresh medium, the Hep-G2 cells were reseeded in tissue culture flasks (Sarstedt, UK).2.3 Preparation of whey protein (WP) particles
Whey protein powder was dissolved in water at a concentration of 2 wt% for 2 h under agitation. The solution was placed at 4 °C for 12 h to fully hydrate the whey protein. Then, the solution was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 × g for 1 h and the supernatant was collected. A solution of 300 mM NaCl was prepared and mixed with an equal volume of WP solution. The pH was adjusted to the required value by drop-wise addition of filtered 0.5 M HCl aqueous solution. After heating the WP/NaCl solution in an oil bath at 85 °C for 15 min, it was left to cool at 4 °C. This precipitation process produced WP particles, which were used in our protocol as stabilizers for the water-in-water Pickering emulsions.
800 × g for 1 h and the supernatant was collected. A solution of 300 mM NaCl was prepared and mixed with an equal volume of WP solution. The pH was adjusted to the required value by drop-wise addition of filtered 0.5 M HCl aqueous solution. After heating the WP/NaCl solution in an oil bath at 85 °C for 15 min, it was left to cool at 4 °C. This precipitation process produced WP particles, which were used in our protocol as stabilizers for the water-in-water Pickering emulsions.
2.4 Production of hepatic cell clusteroids
The cell clusteroid fabrication process was somewhat similar to the procedure described in ref. 23 and 24. Briefly, PEO aqueous solution (5.5 wt%) was prepared by dissolving PEO into the heat-treated solution of WP, which constituted the continuous phase of the water-in-water emulsion. Centrifugation of the PEO solution was done beforehand at 5000 × g for 7 min to remove the silica nanoparticles from the PEO solution, which have been added by the manufacturer. A solution of 5.5 wt% dextran (in total) in EMEM complete medium under sterile conditions was used as a disperse phase (DEX phase) together with the Hep-G2 cells. The DEX phase with the hepatocytes formed different volume fractions with respect to the DEX/PEO w/w emulsion. To form the latter, the DEX phase was mixed with the pelleted cells and then was transferred to the WP/NaCl/PEO solution, and gently homogenized using two pumps with a BD Microlance™ 3, 6 or 12 needles (21G 12, internal diameter 0.512 mm) and a BD Plastipak™ syringe of 5 mL using two pumps (BD biosciences, Wokingham, UK). The w/w emulsion was prepared using PEO and DEX phases at various volume fractions of the two phases. The emulsion was further mixed with an aliquot of PEO solution of higher concentration (14 wt% PEO) to reach a total concentration of 10% in order to osmotically squeeze the DEX droplets with the cells into densely packed clusteroids. The cell clusters were left in the emulsion for 2 h to achieve better cell–cell adhesion and stick together. The emulsion would then be broken down by adding ten times higher volume of EMEM medium, and the cell clusteroids were settled by gravity-driven sedimentation.252.5 3D hepatocyte clusteroids culture
An aqueous solution of 2 wt% sodium alginate was prepared and then sterilized in an autoclave. EMEM complete medium was mixed with the 2 wt% sodium alginate solution at different volumes to vary the gelling agent overall concentration. Hep-G2 cell clusteroids were then carefully resuspended in this solution and seeded on 24-well tissue culture plates. The addition of 2 wt% CaCl2 (aq) allowed the crosslinking of the alginate chains in the media and formation of a hydrogel. After 2 min of incubation, the CaCl2 solution was carefully pipetted out without compromising the integrity of the hydrogel–clusteroid composite and the wells were topped up with EMEM medium. The Ca2+ ions were used for the crosslinking of the alginate hydrogel. The excess CaCl2 solution was removed after the hydrogel formation. The crosslinking process takes no longer than 2 min which had a very marginal impact on the embedded cells.2.6 Optical microscopy
The microstructure of w/w Pickering emulsion at different ϕDEX ratios and the resulting clusteroids at different forms were first analyzed using a fluorescence microscope (Olympus BX51), and samples were observed under various water immersion objectives at ambient temperature (25 °C). Briefly, 30 μL dispersions were shifted and spread onto the hollow glass slide, which was then enclosed by the glass cover.2.7 WP particle size distribution measurement
The WP particle size was measured after diluting (100×) the samples with purified water to an appropriate particle concentration using the Zetasizer Nano ZS (Malvern Instruments Ltd., Malvern, Worcestershire, UK) and Malvern MasterSizer 3000. All measurements were carried out at room temperature and the reported results were the averages of three readings.2.8 Cell viability assay
Cell clusteroids were treated with a 5 g L−1 solution of fluorescein diacetate (FDA, from Sigma-Aldrich) in acetone (10 μL of solution per 1 mL of the dispersion of re-suspended clusteroids) to evaluate cell viability. After 10 min of incubation at room temperature in the dark, the sample was observed using an Olympus BX-51 fluorescence microscope (Olympus) with a DP70 digital camera and FITC fluorescence filter set. FDA is known to diffuse through cell membranes and only viable cells are able to hydrolyze non-fluorescent FDA internally to fluorescein by intracellular esterases. Since fluorescein dissociates in water, its crossing of the cell membranes is hindered by its ionic charge, which results in the accumulation of the fluorescent byproduct inside the intact cells.23,24 This indicates that the cell membranes are intact and the cells are viable.2.9 Urea and albumin production assays
Urea concentration in the samples was quantified using a colorimetric endpoint assay based on acid- and heat-catalyzed condensation of urea with diacetylmonoxime (Stanbio Labs). 100 μL cell culture medium was collected and homogenized with urea assay buffer and analyzed according to the manufacturer's protocol. Due to the possible interference of the alginate gel on the readings for albumin, albumin was tested after the sample was de-gelled. Briefly, 1 mL of 5 mg mL−1 alginate lyase was added to each well followed by 30 min incubation at 37 °C. Albumin content in the de-gelled media was measured using an enzyme-linked immunosorbent assay (ELISA) with horseradish peroxidase detection and peroxidase substrate 3,3,5,5-tetramethylbenzidine as described according to the manufacturer's protocol. The absorbance (450 nm) of the samples was determined using a Synergy HT plate reader and values were quantified from a standard curve. Samples were collected after 72 h of the initial cell seeding from 2D cultures. Changing the culture media every day was essential for optimal growth in 3D cultures and so 24 h cumulative albumin secretion was measured at days 1, 2, 3, 4, 5, 6 and 7.2.10 Cryo-SEM imaging
Cryo-scanning electron microscopy (Cryo-SEM) images of the clusteroids were taken in order to reveal their morphology. Samples were prepared for Cryo-SEM imaging after fixing the cell clusteroids to avoid them breaking apart during the evaporation of the aqueous solution. 1 mL aliquots of the Hep-G2 cell clusteroids in media were deposited on dry Aclar™ sheets (Agar Scientific Ltd, Essex, UK) or poly-lysine coated glass coverslips and treated with 2 wt% glutaraldehyde for 1 h, washed multiple times with Mill-Q water and subjected to critical point drying. The samples were then imaged using a Cryo-scanning electron microscope SEM (ZEISS EVO 60 EP-SEM).2.11 Statistical analysis
Comparisons were made using unpaired t-tests or analysis of variance as appropriate with significance set at p ≤ 0.05.2.12 Crystal Violet staining
The 96-well plates were stained with Crystal Violet (CV) by adding 130 μL to each cell. The 96-well plates were then incubated for 30 min at room temperature and then rinsed 3 times by gentle submersion in water and allowed to dry overnight. After the wells were fully dried, the CV was solubilised by adding 125 μL from a stock solution of 30% acetic acid in deionized water to each cell in the 96-well plates. The solubilised CV solution was then transferred to new 96-well plates and absorbance readings were taken using the Bio-Tek Synergy™ HT Multi-Detection Microplate Reader at 550 nm. The 30% stock solution of acetic acid was used as a blank. The absorbance readings were then normalised.3. Results and discussion
3.1 WP particle characterization
Previous studies showed that emulsions made from preheated and aggregated proteins were more stable than the ones with native proteins.26–28 The heat-treatment allows them to make protein aggregates that would stronger adsorb at the interface between the continuous and dispersed aqueous phases.The whey protein particles are a biocompatible stabilizer of the w/w Pickering emulsions. They are a very essential component in preventing DEX droplets dispersed in the PEO phase from coalescing which leads to emulsion breakdown. The absence of protein particles leads to phase separation in a very short time and due to poor emulsion stability.24,28,29 This makes the results without a particle stabilizer poorly reproducible. In the presence of the whey protein particles, the w/w Pickering emulsions (DEX-in-PEO) had sufficient stability which allowed us to handle the cell-loaded emulsions over the selected time interval and produce cell clusteroids of reproducible average size. Indeed, protein particles with a radius higher than 85 nm have been found to increase droplet size.28 WP particles produced by our methodology were characterized before using them to stabilize the w/w Pickering emulsions. Fig. 2 shows a typical size distribution of the WP particles at different pH. The WP particles showed that the most homogenous peak was at pH 6.18 with an average particle size of 130 ± 30 nm. Consequently, pH 6.18 was chosen for the preparation of WP particles used in the following experiments.
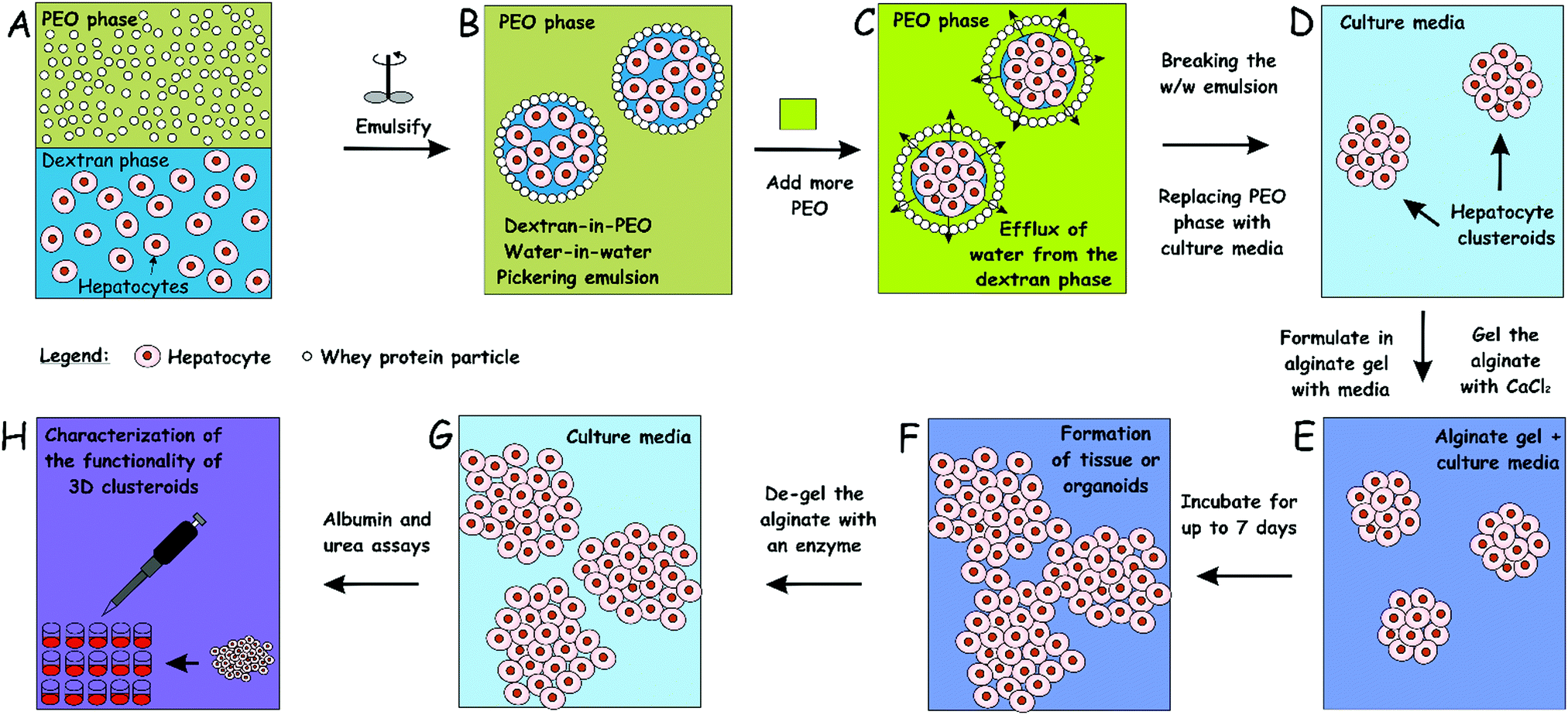 | ||
| Fig. 1 Schematics of our high-throughput method for the preparation of hepatocyte cell clusteroids (A–D) and 3D tissue culturing (D–F) with characterization (G and H). The hepatocyte cells are encapsulated in a dextran-PEO water-in-water emulsion template stabilized by 2 wt% whey protein (WP) particles. The continuous phase is PEO 5.5 wt% and the dispersed phase is composed of cells encapsulated in 5.5% dextran droplets. Upon emulsification, the cells prefer the discontinuous dextran phase, which allows their encapsulation. Adding a more concentrated PEO phase causes osmotic shrinking of the cell-rich dextran drops, whose interfacial tension packs the cells into tissue clusteroids. The latter are isolated by breaking the emulsion by dilution with culture media. | ||
 | ||
| Fig. 2 The distribution of the hydrodynamic diameter of the produced WP particles at different pH. | ||
In our experiments, stable DEX-in-PEO emulsions did not form without the WP particle stabilizers. Thorough research of the DEX/PEO aqueous two phase system stabilized by other protein particles without cell encapsulation has been conducted by Balakrishnan et al.33 These systems show strong stability and could last for days. However, water-in-water Pickering emulsions without cells are irrelevant to this study and behave differently to those with cells.
3.2 Water-in-water emulsion characterization
In order to control the size of the DEX droplets, Pickering w/w emulsions of different volume fractions of the DEX phase were produced: ϕ = 0.1, 0.11, 0.125, 0.14, 0.17, 0.2, 0.25, 0.33 and 0.5. The images shown in Fig. 3 demonstrate that the variation of the DEX phase volume has a strong impact on the emulsion droplet size. The average size of the DEX emulsion droplets with ϕDEX = 0.5 was about 80 μm, with a sharp decrease to 5 μm when the ϕDEX decreased to 0.14. After the shrinking stage, the DEX droplets (without any cells) had a similar size distribution around 5 μm at any ϕ ratio, which demonstrates the strong effect of the osmotic pressure driven by the addition of high concentration PEO solution (Fig. 4). Hence, decreasing the volume fraction of the DEX phase resulted in smaller DEX droplets with the decreased capacity for encapsulating a lower number of cells. The microscopy observation of DEX droplets shown in Fig. 3, showed that the water-in-water emulsions contain a higher drop size with ϕDEX = 0.2 and 0.17. Consequently, ϕDEX = 0.2 was selected for use in our production of hepatocyte clusteroids in this research.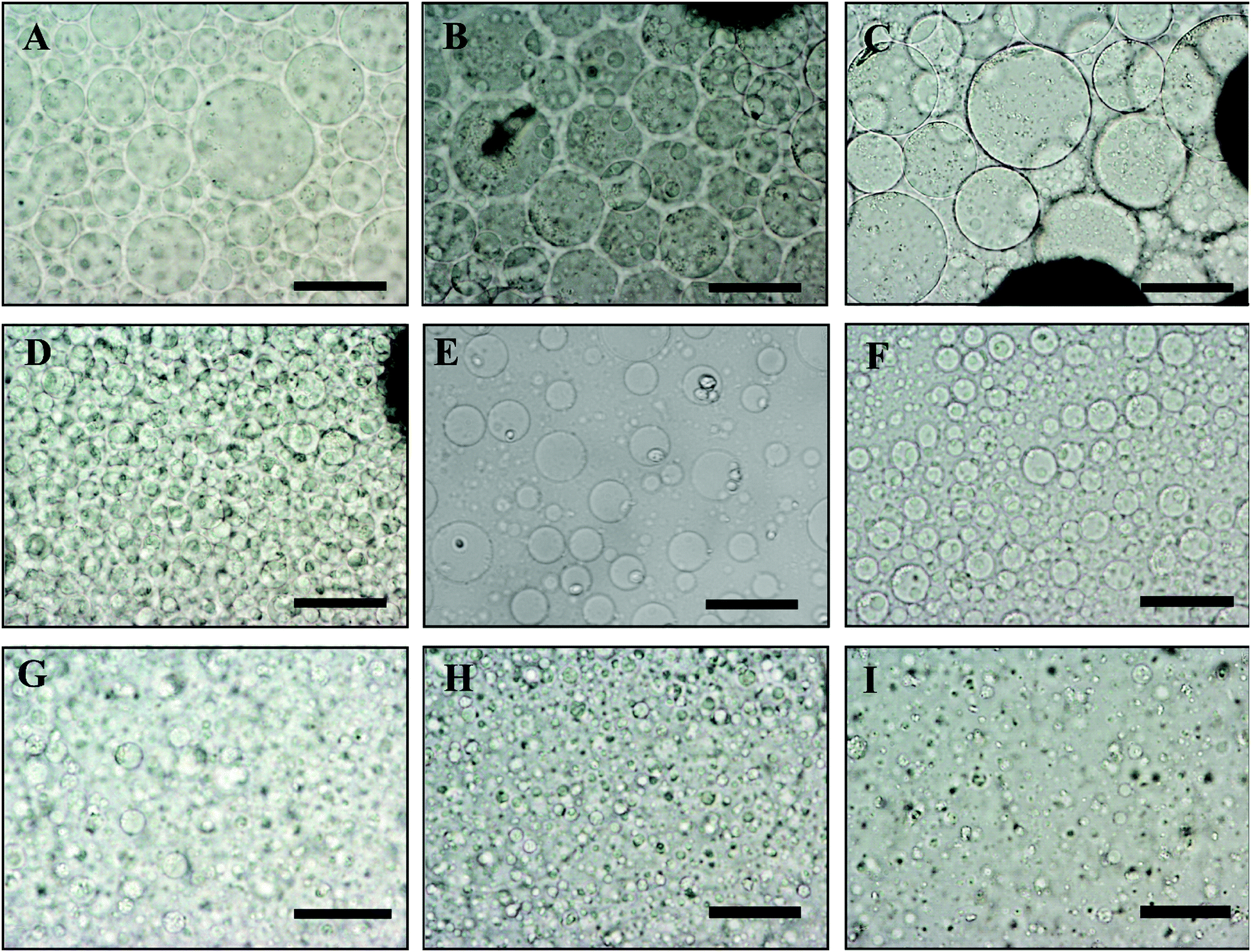 | ||
| Fig. 3 Optical microscopy images of a DEX/PEO water-in-water Pickering emulsion (PEO 5.5 wt% and DEX 5.5 wt%). (A) ϕDEX = 0.5, (B) ϕDEX = 0.33, (C) ϕDEX = 0.25, (D) ϕDEX = 0.2 (E), ϕDEX = 0.17 (F), ϕDEX = 0.14, (G) ϕDEX = 0.125, (H) ϕDEX = 0.11 and (I) ϕDEX = 0.1, stabilized by 2 wt% WP particles at pH 6.18. Scale bars are 100 μm. | ||
 | ||
| Fig. 4 Average DEX droplet diameter for the DEX/PEO Pickering emulsion produced from WP/NaCl 300 mM solution at pH 6.18, 5.5 wt% PEO/5.5 wt% dextran for varying volume fractions of the DEX phase (average of 200–300 individual drops). The average drop diameters are represented before shrinking (green bars) and after shrinking (blue bars). The data were obtained by optical microscopy measurements of the DEX droplets for each micrograph with ImageJ software. | ||
3.3 Effect of the hepatic cells’ encapsulation on the average drop size of the w/w Pickering emulsion
For the sake of optimization and potential control of the clusteroids formation, the effect of the cell encapsulation on w/w Pickering emulsion drop size was further characterized. To prepare the w/w emulsion with encapsulated cells, HepG2 cells were mixed with a DEX-based solution made with EMEM media. The resulting mixture was added to the PEO-based solution, followed by gentle mixing using a syringe as described earlier. One can see from Fig. 5 and 6 that the cells packed in the DEX droplets slightly increased the DEX droplet diameter. Meanwhile, there remain cavities where the DEX droplets had not fully shrank; in stark contrast, the clusteroids were densely packaged in DEX droplets after the contraction. The scale of the contraction is about 25%, from a diameter of 35 μm to 24 μm. Hence the effect of the osmotic pressure is strong enough to reduce the drop size and their interfacial tension to compress the encapsulated cells into spherical clusters, thus increasing their natural cell–cell adhesion as they gain more contact area with each other. | ||
| Fig. 5 Average DEX droplet diameter for the DEX/PEO Pickering emulsion produced from WP/the average diameter of Hep-G2 cell clusteroids and drops with encapsulated hepatic cells. The data were obtained by optical microscopy measurements of 100–200 individual clusteroids for each micrograph with ImageJ Software (Student's t-test, NS: non-significant, **p < 0.01, ***p < 0.001). | ||
 | ||
| Fig. 6 Optical microscopy images of (A, B, E, F) Hep-G2 cell-loaded DEX/PEO emulsion droplets (before shrinkage) and (C, D, G H) Hep-G2 cell clusteroids obtained (after shrinkage of the DEX phase). The initial w/w emulsion was stabilized by 2 wt% WP particles with 5.5 wt% PEO/5.5 wt% Dextran. Here the DEX volume fraction is ϕDEX = 0.2. Scale bars are 100 μm for (A, B, E, F) and 50 μm for (C, D, G, H). | ||
3.4 Cell clusteroid formation in DEX-in-PEO emulsions
Three-dimensional cultures of tumour cells have recently gained increasing interest in cancer research. These spheroids are three-dimensional clusters of tumour cells that mimic various properties of solid tumours and present a relevant tumour model for drug discovery.11,29–31The dependence of the cell spheroid's size on the volume fraction of DEX/PEO has been studied before by Celik et al.23 for HaCaT cells. Their results show that the spheroid's size depends not only on the DEX/PEO ratio but also on the cell concentration in the APTS system. Given the complexity of such phase diagram and the dependence of the obtained spheroid's size on the cell type and concentration, it was not practical to scan the whole range of parameters. The clusteroid's size is also sensitive to the osmotic shrinking procedure, i.e. the addition of the concentrated PEO phase. Therefore, we adopted the optimized protocol by Celik et al.23 for the preparation of clusteroids with hepatic cells and the current focus of this work is to demonstrate the enhanced functionality of clusters.
To demonstrate the efficiency of the cell encapsulation, we performed a spheroid assay using ATPS system made with PEO (MW 35 kDa)–DEX (MW 500 kDa) solutions. A suspension of Hep-G2 cells was generated, and this suspension was dispensed and mixed with PEG phase solution. Because the density of cells was between that of the DEX and the PEO solution, the cells accumulated at the interface of the two phases after 1 h, as shown in Fig. S2 (ESI†). The collected clusteroids were imaged using the microscope, as shown in Fig. 7, and seem compatible in structure and relatively uniform in size, at about 20 μm. The osmotic pressure compacted the cells in a spherical shape and makes them have better cell–cell adhesion, which helps the formation of the clusteroids.
 | ||
| Fig. 7 Optical microscopy images of Hep-G2 cell droplets encapsulated in the DEX/PEO water-in-water emulsions after shrinkage stabilized by 2 wt% WP particles at different magnifications. Scale bars are (A, B, D, E) 100 μm and (C, F) 50 μm. | ||
Our results showed that Hep-G2 cells can form compact clusteroids within 1 h of incubation in the w/w emulsions. Therefore, we evaluated spheroid formation with all four two-phase solutions after 1 h. Next, we examined the viability of the longer-term culture. Viability of samples of the hepatic cell spheroids and the single cells was initially tested using the FDA live/dead assay before being added to the gel. One can see from Fig. 8 that the viability of the individual (single) cell and the one in the clusteroids are both high. This demonstrates that the viability of the cells in the clusteroids is not affected by our preparation method. These results combined with the data obtained with water-in-water emulsion showed that ϕDEX= 0.2 and 10 wt% PEO as a DEX drop shrinking concentration are suitable for the preparation of Hep-G2 cell clusteroids and this does not impair their further growth. This composition was used for the following study.
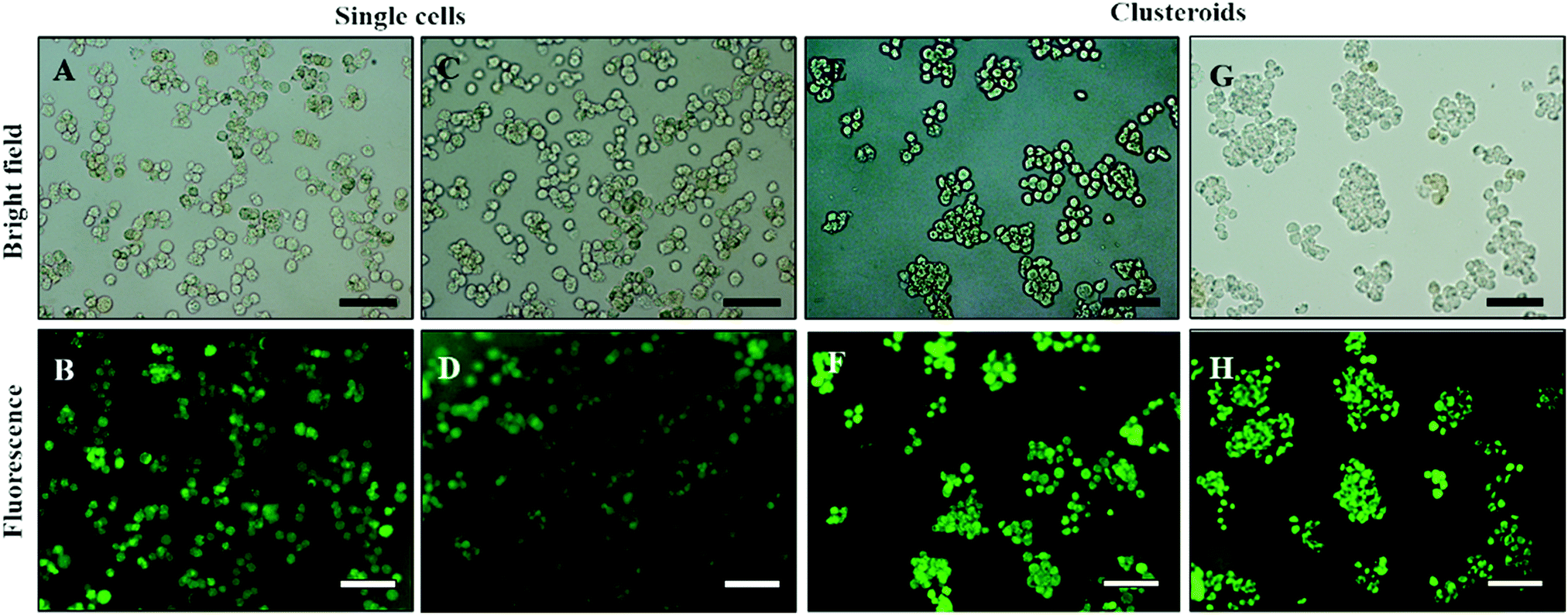 | ||
| Fig. 8 (A and C) Optical bright-field microscopy images and (B and D) fluorescence microscopy images of single Hep-G2 cells after being treated with the FDA live/dead assay. (E and G) Optical bright-field images and (F and H) fluorescence microscopy images (F and H) of Hep-g2 cell clusteroids treated with 1 mM FDA. FDA treatment was done after the cell clusteroid fabrication. Green fluorescence indicates that both the Hep-G2 cells preserve their viability during the clusteroid fabrication process (Fig. 1). The scale bar is 100 μm. | ||
3.5 Hepatic clusteroid growth during long-term culture
We examined the compatibility of the assay with longer-term culture. Clusteroids of two different cell densities of 1 × 106 to 1 × 105 mL−1 were generated within 50 μL DEX drops immersed in the PEO phase and then put into alginate hydrogel fortified by 1 M CaCl2 treatment as described earlier. Each condition was set with 12 replicates, that is, half of a 24-well plate. After every 24 hours of growing the clusteroids within each well, we added 1 mL of the same growth medium to the wells after withdrawal of the same amount of old medium. Therefore, the collected urea and albumin produced by the clusteroids each day was removed from culture, and not accumulated. Clusteroids were imaged every other day. Fig. 9 contains images showing that after three days of their growth, the cells change from a cluster of individual cells to thicker, globular structures. The clusteroids percolated throughout the hydrogel to make contacts with cells from neighbouring clustered cells after 5 days of culturing. We were unable to further monitor the clusteroids’ diameter beyond seven days due to the percolation of the hepatocyte clusteroids and the formation of denser tissues (see Fig. 9). | ||
| Fig. 9 Bright-field optical microscopy images of (A, B, C, D) monolayer cultured single Hep-G2 cells (E, F, G, H) Hep-G2 clusteroids isolated by dilution of the DEX/PEO emulsion by a factor of 3 with EMEM medium and incorporated with 1 wt% sodium alginate in EMEM media followed by cross-linking with 1M CaCl2. The Hep-G2 cell clusteroids were cultured in the alginate film for seven days in EMEM media and images were taken from each well to determine the average clusteroid size. Scale bars are 100 μm. | ||
As shown in Fig. 9, the growing mode of single cells and clustered cells is completely different. The single cells proliferated individually and would not form a cluster or spheroid structure. In contrast, the clusteroids we imaged in the alginate gels grew to form a compact tissue structure. The result clearly shows that the alginate hydrogel does not provide a suitable environment for clusteroid formation. The clusteroid formation in our experiments is the result of the directed templating of the w/w Pickering emulsion droplets on the embedded cells. This result indicates that our technique can make the clustered cells grow in the pattern in which the cell would proliferate surrounding the initial core.
The clusteroids’ size was measured manually using ImageJ software as the cell clusteroids continued growing in the seven day culture (Fig. 10A). The cell size increased strongly in the first three days, from 24 μm to 41 μm, and then increased slowly to 55 μm in the following 4 days, which could be attributed to the larger number of cells required to enlarge the cell clusteroids. Here we aimed at using the produced clusteroids in tissue engineering applications which requires a high yield of clusteroids instead of absolute size uniformity. The clusteroid uniformity is essential when using them for drug testing. For growth monitoring, the clusteroids with lower cell numbers of 1 × 105 mL−1 were also put in the alginate gels to observe the individual clusteroid growth.
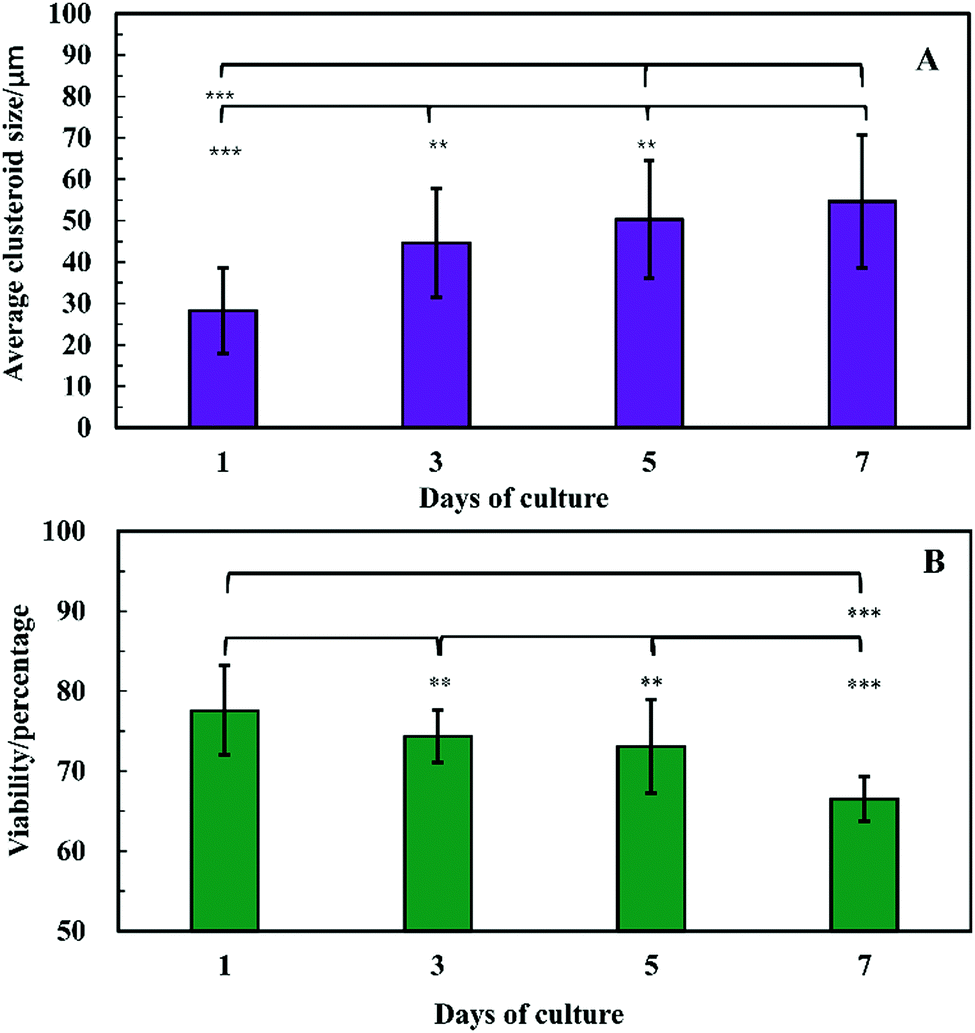 | ||
| Fig. 10 Evolution of the Hep-G2 clusteroids embedded in a hydrogel composed at different days of culture. (A) The spheroid size vs. time and (B) viability vs. time. Measurements of the clusteroids were taken every day with ImageJ software by taking the average of the vertical and horizontal diameter of 500 clusteroids in each well. (**p < 0.01,***p < 0.001). | ||
In order to examine the viability of the clusteroids, samples were taken out from the alginate gel by adding 5 mg mL−1 alginate lyase solution. The viability of the cells in the clusteroids was characterized on different days. The cells' viability decreased over a few days of culture, from 78% to 65% (Fig. 10B). This might be the result of necrotic core formation when the clusters become larger. The microscopic observations of the FDA treated clusteroids after various days are shown in Fig. S3 (ESI†).
One can see from Fig. S4 (ESI†), that after 3 days of culture in a hydrogel, the cell clusteroids became dense and compact. A single Hep-G2 clusteroid could reach up to 80 μm after seven days’ culture. SEM images of samples of such grafts after freeze-drying at a critical point are shown at different resolutions in Fig. 11. The clustering of the clusteroids in the hydrogel matrix and the way they have started to spread out and percolate in the gel should be noted. This process led to the formation of integral tissues over the course of seven days. Interestingly, the cell clusteroids had a relatively uniform size. The clusteroids form a dense layer which is highly desirable for tissue engineering applications.
 | ||
| Fig. 11 SEM images of a sample of Hep-G2 cell clusteroids encapsulated in sodium alginate gels after 7 days of culture, deposited on a glass substrate and freeze-dried before imaging. (A–C) Images correspond to different resolutions. Note that the size of the clusteroids of cells is slightly lower than the original cell clusteroids due to shrinkage. The bars are (A) 100 μm; (B) 10 μm; (C) 1 μm. | ||
3.6 Cell clusteroids liver-specific functionality
Cell polarity and ECM signalling have been shown to promote liver-specific functions such as albumin secretion and metabolic enzyme expression.32 To study the liver-specific functions of the clustered Hep-G2 cells, albumin secretion and urea synthesis by 1 × 106 of HepG2 cells pre-clustered in clusteroids, cultured in series of 1 wt% sodium alginate gel, were quantified. Urea synthesis was quantified over 7 days of culture. Fig. 12A shows the amount of albumin secreted into the medium per day in each well after fixed number of days of accumulation. A trend was found for cells cultured with gels: the albumin secretion increases with longer culturing. In alginate gel cultured with single cells, albumin secretion was slightly higher than the gels encapsulated with clustered cells at day 1. The result shown in Fig. S5 (ESI†) could also demonstrate that the albumin production increased for every 24 h in culture. On day 10, the amount of albumin secreted by Hep-G2 clustered cells was approximately three times higher than that of similar number of individual Hep-G2 cells. The result suggests that albumin secretion of the cells is improved in clusteroids. Urea synthesis is another important liver-specific function of Hep-G2 cells. The urea production profile of the cells was similar to that for albumin secretion. As shown in Fig. 12B, after 7 days of culture, urea synthesis by the accumulation of both the individual cells and clusteroids increases with time, but the urea production per day was continuously decreasing for the seven days as shown in Fig. S5B (ESI†). | ||
| Fig. 12 (A) Urea synthesis and (B) albumin secretion by Hep-G2 cells in blend gels as a function of culture time by accumulation. Data are shown as mean ± standard deviation from the two samples. The superscript letters represent a significant difference between groups (**p < 0.01, ***p< 0.001). | ||
This could be attributed to the reduction of mesothelial cells with the cell proliferation, which accounts for urea production.2 In addition, the urea synthesis rate by the clusteroids was two times higher than the single cells, confirming again that the introduction of clusteroids enhances the liver-specific functions of the Hep-G2 cells. The result indicates that the cell–cell interactions in these clusteroids benefit their high level of liver-specific functions as revealed in Fig. 12.
4. Conclusions
3D cultures of tumor cells as spheroids present a physiologically relevant in vitro model that captures some key features of solid tumors and could prove to be an essential tool for compound screening to identify potent anti-cancer drugs. Despite these recognized advantages, spheroids remain underutilized in high throughput screening applications due to the limitations of the existing preparation techniques. We present a new approach for the 3D culture of hepatic tumor cells (Hep-G2) using immiscible aqueous solutions of two biopolymers, dextran and PEO. We show that water-in-water Pickering emulsion droplets of the dextran aqueous phase containing the Hep-G2 cells encapsulate the cells and can facilitate the cell aggregation and formation of compact cell spheroids by increasing the osmotic pressure of the continuous phase of PEO solution. This was done by adding more PEO to the continuous phase after the emulsion is formed. The cell clusters that are obtained from each DEX drop after osmotic shrinking with PEO were harvested after breaking down the emulsion by dilution with culture media. This approach allows the formation of consistent tissues in standard 24-well plates, containing thousands of spheroids, to allow further biochemical assays of cell responses in situ using plate readers. We incorporated the hepatic cell clusteroids in alginate gel and followed their growth over a course of up to 7 days compared with the same number of single cells under the same conditions. The liver-specific function of the hepatocytes was greatly enhanced due to the formation of cell clusteroids. This is an essential feature for application in tissue engineering as the same initial number of cells grow faster, as well as for efficient high throughput screening applications or further drug testing platforms. This approach to the 3D culture of hepatic cells will benefit a broad range of applications in tissue engineering and drug library screening without a need for special plates, devices, and facilities.Author contributions
V. N. P. envisaged the work and supervised the project team. V. N. P., L. A. M. and co-supervised A. W. and provided technical advice. A. W. worked on the cell culturing, preparation of the clusteroids and the tissue engineering with them. A. W. prepared the draft manuscript which was then co-edited and completed through contributions of all authors. All authors have given approval to the final version of the manuscript.Funding sources
A. W. is grateful to the Chinese Scholarship Council for the financial support of this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors appreciated the technical help from Tony Sinclair and Ann Lowry at the University of Hull Microscopy Suite with the SEM sample preparation and imaging. A. W. acknowledges the Chinese Scholarship Council for his PhD studentship.References
- X. Gu and J. E. Manautou, Expert Rev. Mol. Med., 2012, 14, e4 CrossRef.
- J. A. Kyffin, P. Sharma, J. Leedale, H. E. Colley, C. Murdoch, A. L. Harding, P. Mistry and S. D. Webb, Toxicol. In Vitro, 2019, 55, 160–172 CrossRef CAS.
- N. G. McDowell, R. A. Fisher, C. Xu, J. C. Domec, T. Holtta, D. S. Mackay, J. S. Sperry, A. Boutz, L. Dickman, N. Gehres, J. M. Limousin, A. Macalady, J. Martinez-Vilalta, M. Mencuccini, J. A. Plaut, J. Ogee, R. E. Pangle, D. P. Rasse, M. G. Ryan, S. Sevanto, R. H. Waring, A. P. Williams, E. A. Yepez and W. T. Pockman, New Phytol., 2013, 200, 304–321 CrossRef CAS.
- M. Eichelbaum, M. Ingelman-Sundberg and W. E. Evans, Annu. Rev. Med., 2006, 57, 119–137 CrossRef CAS.
- K. M. Lee, D. Kang, M. L. Clapper, M. Ingelman-Sundberg, M. Ono-Kihara, C. Kiyohara, S. Min, Q. Lan, L. Le Marchand, P. Lin, M. L. Lung, H. Pinarbasi, P. Pisani, P. Srivatanakul, A. Seow, H. Sugimura, S. Tokudome, J. Yokota and E. Taioli, Cancer Epidemiol., Biomarkers Prev., 2008, 17, 1120–1126 CrossRef CAS.
- L. Yuan and N. Kaplowitz, Clin. Liver Dis., 2013, 17, 507–518 CrossRef.
- S. Blach, S. Zeuzem and M. Manns, Lancet Gastroenterol. Hepatol., 2017, 2, 161–176 CrossRef.
- D. Azoulay, D. Castaing, R. Adam, E. Savier, V. Delvart, V. Karam, B. Y. Ming, M. Dannaoui, J. Krissat and H. Bismuth, Ann. Surg., 2001, 233, 565–574 CrossRef CAS.
- N. Lebonvallet, N. Boulais, C. Le Gall, U. Pereira, D. Gauche, E. Gobin, J. O. Pers, C. Jeanmaire, L. Danoux, G. Pauly and L. Misery, Exp. Dermatol., 2012, 21, 156–158 CrossRef.
- E. Nicholas, S. D. Timmins and L. K. Nilesen, Angiogenesis, 2004, 7, 97–103 CrossRef.
- Y. C. Tung, A. Y. Hsiao, S. G. Allen, Y. S. Torisawa, M. Ho and S. Takayama, Analyst, 2011, 136, 473–478 RSC.
- E. Atefi, S. Lemmo, D. Fyffe, G. D. Luker and H. Tavana, Adv. Funct. Mater., 2014, 24, 6509–6515 CrossRef CAS.
- H. Ota, R. Yamamoto, K. Deguchi, Y. Tanaka, Y. Kazoe, Y. Sato and N. Miki, Sens. Actuators, B, 2010, 147, 359–365 CrossRef CAS.
- G. R. Souza, J. R. Molina, R. M. Raphael, M. G. Ozawa, D. J. Stark, C. S. Levin, L. F. Bronk, J. S. Ananta, J. Mandelin, M. M. Georgescu, J. A. Bankson, J. G. Gelovani, T. C. Killian, W. Arap and R. Pasqualini, Nat. Nanotechnol., 2010, 5, 291–296 CrossRef CAS.
- M.-L. Brandy, O. J. Cayre, R. F. Fakhrullin, O. D. Velev and V. N. Paunov, Soft Matter, 2010, 6, 3494–3498 RSC.
- R. F. Fakhrullin, M.-L. Brandy, O. J. Cayre, O. D. Velev and V. N. Paunov, Phys. Chem. Chem. Phys., 2010, 12, 11912–11922 RSC.
- L. Gasperini, J. F. Mano and R. L. Reis, J. R. Soc., Interface, 2014, 11, 20140817 CrossRef.
- E. Atefi, R. Joshi, J. A. Mann and H. Tavana, ACS Appl. Mater. Interfaces, 2015, 7, 21305–21314 CrossRef CAS.
- S. Singh and H. Tavana, Front. Chem., 2018, 6, 379 CrossRef.
- S. Lemmo, E. Atefi, G. D. Luker and H. Tavana, Cell. Mol. Bioeng., 2014, 7, 344–354 CrossRef CAS.
- S. L. Ham, E. Atefi, D. Fyffe and H. Tavana, J. Vis. Exp., 2015, 98, e52754 Search PubMed.
- P. Shahi Thakuri, S. L. Ham, G. D. Luker and H. Tavana, Mol. Pharm., 2016, 13, 3724–3735 CrossRef CAS.
- S. B. G. Celik, S. R. Dominici, B. W. Filby, A. A. K. Das, L. A. Madden and V. N. Paunov, Biomimetics, 2019, 4, 50 CrossRef CAS.
- A. A. K. Das, B. W. Filby, D. A. Geddes, D. Legrande and V. N. Paunov, Mater. Horiz., 2017, 4, 1196–1200 RSC.
- C. Han, S. Takayama and J. Park, Sci. Rep., 2015, 5, 11891 CrossRef.
- B. P. Binks and H. Shi, Langmuir, 2019, 35, 4046–4057 CrossRef CAS.
- N. Chatsisvili, A. P. Philipse, B. Loppinet and R. H. Tromp, Food Hydrocolloids, 2017, 65, 17–23 CrossRef CAS.
- B. T. Nguyen, T. Nicolai and L. Benyahia, Langmuir, 2013, 29, 10658–10664 CrossRef CAS.
- J. Fukuda, A. Khademhosseini, Y. Yeo, X. Yang, J. Yeh, G. Eng, J. Blumling, C. F. Wang, D. S. Kohane and R. Langer, Biomaterials, 2006, 27, 5259–5267 CrossRef CAS.
- Y. Wang and J. Wang, Analyst, 2014, 139, 2449–2458 RSC.
- Y. Li and E. Kumacheva, Sci. Adv., 2018, 4, 1–11 Search PubMed.
- S. C. Ramaiahgari, M. W. den Braver, B. Herpers, V. Terpstra, J. N. Commandeur, B. van de Water and L. S. Price, Arch. Toxicol., 2014, 88, 1083–1095 CAS.
- G. Balakrishnan, T. Nicolai, L. Benyahia and D. Durand, Langmuir, 2012, 28, 5921–5926 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: In the enclosed electronic supplementary information (ESI), we present the following additional data: (i) droplet shrinking effect of adding PEO to the w/w Pickering emulsion; (ii) schematics of the formulation of Hep-G2 clusteroids; (iii) clusteroids’ viability at different days of culture; (iv) long-term culture of clusteroids in the hydrogel matrix; (v) urea and albumin production of hepatocyte clusteroids at different days of culture. See DOI: 10.1039/d0ma00635a |
| This journal is © The Royal Society of Chemistry 2020 |