
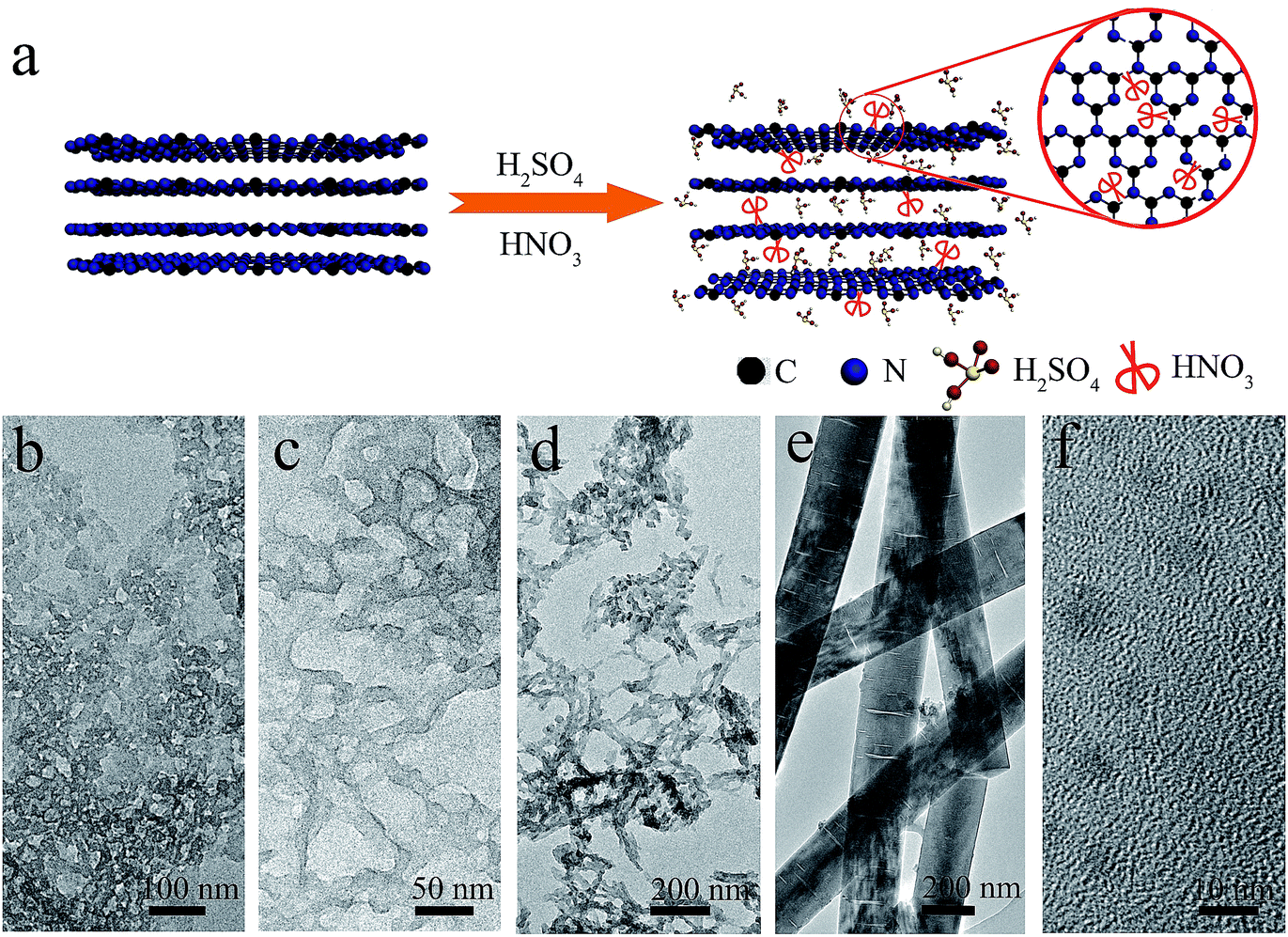
Graphitic carbon nitride nanoribbon for enhanced visible-light photocatalytic H2 production†
Xiuming
Bu
ab,
Yu
Bu
a,
Siwei
Yang
b,
Feng
Sun
a,
Linfan
Tian
b,
Zheng
Peng
b,
Peng
He
b,
Jing
Sun
b,
Tao
Huang
b,
Xianying
Wang
*a,
Guqiao
Ding
*b,
Junhe
Yang
a and
Xiaoming
Xie
b
aSchool of Materials Science and Engineering, University of School for Science and Technology, Shanghai 20093, P. R. China. E-mail: xianyingwang@usst.edu.cn
bSchool Key Laboratory of Functional Materials for Informatics, Shanghai Institute of Microsystem and Information Technology, Chinese Academy of Science, Shanghai 200500, P. R. China. E-mail: gqding@mail.sim.ac.cn
First published on 17th November 2016
Abstract
Chemical scissors provide a new vision to manufacture unique carbon nitride nanostructures with improved photocatalytic performance. Herein, graphitic carbon nitride nanoribbon (GCNR), with a typical length of 1.75 μm, width of 210 nm and thickness of 3 nm, was obtained by acid treatment of bulk g-C3N4 with HNO3/H2SO4 mixtures. The C/N molar ratio of GCNR (around 0.629) was much smaller than that of pristine g-C3N4 (0.758). It was demonstrated that larger amounts of carbon vacancies on the ultra-thin ribbon structures could contribute to improved electron–hole separation efficiency and excellent photocatalytic H2 production. The average H2 production rate of GCNR under visible light was 49.4 μmol h−1, which was 20 times that of the original catalyst.
Development of highly active and green photocatalysts for environmental protection and energy shortage has attracted lots of attention during recent years.1,2 Among various photocatalysts, graphitic carbon nitride (g-C3N4), as a non-metal organic semiconductor, has been deeply and widely researched due to its low cost, excellent anti-photocorrosion and chemical stability.3 However, bulk g-C3N4 directly synthesized from thermal polymerization usually has the disadvantages of low specific area, less active sites and fast electron–hole combination rate, thus hinders their photocatalytic performance.4 Meanwhile, serious aggregation and restacking can be caused due to the π–π stacking and van der Waals attraction between g-C3N4 nanosheets, and thus limit the transport of H2O2 intermediate product which usually poisons g-C3N4 nanosheets during the H2 production process.3a
Various solutions towards solving these problems of bulk g-C3N4 have been developed, which mainly include two approaches: texture modification and control of polymerization degree.5 The structure and composition of carbon nitrides could be engineered by doping of non-metal/metal elements, copolymerization or morphology control. Previous works have shown that special structures, i.e., nanoparticle,6 nanosheet,7 nanorod,8 hollow microspheres9 and porous materials,10 can not only endow them with numerous boundaries and thus decrease the aggregation, but more importantly can guide charge movement to accelerate the collection and separation of electron–hole pairs at materials interface for efficient utilization of solar energy to drive relevant reactions.11 Dong et al. prepared high-quality C3N4 nanosheet with alkali treatment and inorganic salt assistance method respectively which both show excellent photocatalytic activity.12 Yang et al. has reported free-standing g-C3N4 nanosheets prepared by liquid phase exfoliation have large aspect ratios, high surface area, and stoichiometric N/C ratio.13 Further treatment by increasing the amounts of active sites on the g-C3N4 structures would also improve the photocatalytic performance.14 Hong et al. showed that the nitrogen-deficient g-C3N4 synthesized by hydrothermal treatment with oxidant owned an enhanced photocatalytic H2 evolution rate.15 Therefore, it is very meaningful to make active vacancies on the 2D g-C3N4 nanosheet for enhancing photocatalytic performance.
In this work, a facile way was developed to synthesize g-C3N4 nanoribbon (GCNR) by acid treatment (H2SO4/HNO3) of pristine g-C3N4 (PCN). Meanwhile, g-C3N4 was exfoliated into nanoribbons with abundant in-plane carbon vacancies due to strong chemical exfoliation and etching effect. The abundant carbon vacancies may be beneficial for hindering the recombination of electrons and holes when they provide trapping sites for photoinduced holes. The result is photoluminescence and time-resolved fluorescence decay spectra demonstrated that the charge life time and separation of photoinduced electron–hole of GCNR structures have been greatly improved compared to the PCN. Moreover, the hydrogen evolution rate was improved from 2.5 to 49.4 μmol h−1 under visible light.
The volume ratio between H2SO4 and HNO3 was crucial to the formation of nanoribbon structures, and the optimised volume ratio of H2SO4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 was 1:2. Fig. 1a is the transmission electron microscopy (TEM) images of the GCNR. The nearly transparent feature of the samples indicated the ultra-thin feature. The length distribution of as-prepared GCNR was shown in the inset of Fig. 1, which was evaluated from the SEM images by casually selecting 80 nanoribbons (Fig. S1†). The length ranged from 1 to 3 μm, with the average length of 1.7 μm. The width of nanoribbons is about 210 nm. The pores on the surface of the GCNR may be resulted from the strong irradiation of electron-beam. Atomic force microscopy (AFM) was used to measure the thickness of nanoribbons. Two nanoribbons were chosen randomly and their height profiles were around 2.9 nm and 3.1 nm, respectively, which confirmed the ultrathin feature of the GCNR.16 Acid treatment, could exfoliate the layer structures, thus reducing the thickness of PCN.17
:HNO3 was 1:2. Fig. 1a is the transmission electron microscopy (TEM) images of the GCNR. The nearly transparent feature of the samples indicated the ultra-thin feature. The length distribution of as-prepared GCNR was shown in the inset of Fig. 1, which was evaluated from the SEM images by casually selecting 80 nanoribbons (Fig. S1†). The length ranged from 1 to 3 μm, with the average length of 1.7 μm. The width of nanoribbons is about 210 nm. The pores on the surface of the GCNR may be resulted from the strong irradiation of electron-beam. Atomic force microscopy (AFM) was used to measure the thickness of nanoribbons. Two nanoribbons were chosen randomly and their height profiles were around 2.9 nm and 3.1 nm, respectively, which confirmed the ultrathin feature of the GCNR.16 Acid treatment, could exfoliate the layer structures, thus reducing the thickness of PCN.17
 | ||
| Fig. 1 (a) TEM images of the as-prepared PRC; inset of (a) is the size distribution of the sample evaluated from the TEM image. (b and c) AFM image and height profile of the PRCN. High resolution (d) C 1s and (e) N 1s XPS spectra of g-C3N4 and GCNR, respectively. | ||
The N2 adsorption–desorption isotherms of the products were shown in Fig. S2.† The samples exhibited type III isotherms, indicative of mesoporous materials.14 The Brunauer–Emmett–Teller was 2 times larger than that of PCN (11 m2 g−1). The improved specific surface area could facilitate mass transfer during the photocatalytic reaction, possibly enhancing the reaction kinetics.
X-ray diffraction (XRD) was employed to identify the crystal structure and chemical structure of the g-C3N4 before and after treatment. In the XRD patterns (Fig. S3†), the diffraction peak (100) around 13.0° was related to the inter-plane structure packing, while (002) peak at 27.2° indicated the periodic interlayer stacking of conjugated aromatic systems.18 Compared with the PCN, the intensity of GCNR was much lower which indicated the layered g-C3N4 has been successfully exfoliated. This was consistent with the AFM analysis. The valance states and chemical compositions were studied via X-ray photoelectron spectroscopy (XPS). The presence of C, N and O elements can be observed in the survey XPS spectra (Fig. S4†). The corresponding C/N molar ratio of GCNR (0.629) was much smaller than that of pristine g-C3N4 (0.758), indicating the presence of carbon vacancies in GCNR (Table 1). The formation of carbon vacancies may be resulted from the strong acid etching of the g-C3N4 framework. The high-resolution C 1s spectrum was shown in Fig. 2a. The peak centered at 287.8 eV belongs to C–N coordination of g-C3N4, whereas the peak at 288.6 eV is assigned to N–C–N and peak at 284.6 eV is C–C coordination in graphite.19 Interestingly, the peak at 284.6 eV obviously becomes weaken after treatment. The C(C–C)/Ntotal atomic ratio decreases from 0.15 to 0.04. Meanwhile, the deconvolution of high resolution N 1s XPS profiles show three peaks at 398.8 eV, 399.7 eV, 404.7 eV, representing nitrogen in the form of N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N, N–(C)3 and N–H, respectively.11,16 It was found that the atomic percentages of N–H and NC–N bonds decreases slightly may result from the improvement of the groups on the edge after tailored by HNO3. Fig. S5† shows the high-resolution O 1s spectrum where the peaks located at about 531.4 eV and 532.6 eV corresponded to the –OH and CO, respectively. The area ratios of the peak –OH at GCNR are obviously higher than that of PCN indicating that a greater more hydroxyl content on the surface.20 The fourier transform infrared (FTIR) spectra confirms the existence of hydroxyl group after acid treatment as shown in Fig. S6.†
C–N, N–(C)3 and N–H, respectively.11,16 It was found that the atomic percentages of N–H and NC–N bonds decreases slightly may result from the improvement of the groups on the edge after tailored by HNO3. Fig. S5† shows the high-resolution O 1s spectrum where the peaks located at about 531.4 eV and 532.6 eV corresponded to the –OH and CO, respectively. The area ratios of the peak –OH at GCNR are obviously higher than that of PCN indicating that a greater more hydroxyl content on the surface.20 The fourier transform infrared (FTIR) spectra confirms the existence of hydroxyl group after acid treatment as shown in Fig. S6.†
| C 1s | N 1s | ||||||
|---|---|---|---|---|---|---|---|
| Position (eV) | Assignment | C/Ntotal atomic ratio | Position (eV) | Assignment | N/Ntotal atomic ratio | C/N atomic ratio | |
| PCN | 284.6 | C–C | 0.15 | 398.9 | NC–N |
0.69 | 0.758 |
| 287.8 | C–NHX | 0.34 | 400.6 | N–(C)3 | 0.26 | ||
| 288.6 | N–C–N | 0.26 | 404.6 | N–H | 0.05 | ||
| GCNR | 284.6 | C–C | 0.04 | 398.9 | NC–N |
0.65 | 0.629 |
| 287.8 | C–NHX | 0.37 | 400.6 | N–(C)3 | 0.27 | ||
| 288.6 | N–C–N | 0.22 | 404.6 | N–H | 0.08 | ||
 | ||
| Fig. 2 (a) The evolution process of morphology for tailored g-C3N4. (b–f) TEM images of different mixed acid volume ratios: (b) HNO3:H2SO4 = 1:3 (c) HNO3:H2SO4 = 1:2 (d) HNO3:H2SO4 = 1:1 (e) HNO3 : H2SO4 = 2:1 (f) HNO3:H2SO4 = 3:1. | ||
The formation mechanism of GCNR was proposed too. Based on the above experimental data and analysis, the key factor to obtain nanoribbon is to control the volume ratio of HNO3 to H2SO4 accurately. Samples with different volume ratio of HNO3 to H2SO4 are prepared as shown in Fig. S7.† In this process, we believe H2SO4 is responsible for chemical exfoliation while HNO3 results in the structure shape. Previous study has demonstrated the bulk g-C3N4 can be treated by H2SO4 to achieve intercalation and exfoliation.25 After the chemical exfoliation by H2SO4. HNO3 acts as the chemical scissor, carbon atom has a bigger surface area and smaller molecular weight than nitrogen atom, which is prone to break the trizaines (C3N3) first due to heptazines unit (C6N7) is more energetically stable than triazines unit. At the same time, this selective cutting results in the porous structure formation of g-C3N4.26 As the concentration of HNO3 increases, the structure of g-C3N4 can be orientational tailored into nanoribbons. Finally, nanodots can be obtained when the ratio of HNO3:H2SO4 exceeds 3. As seen in Fig. 2, we can observe that when HNO3:H2SO4 volume ratio less than 2, porous g-C3N4 can be obtained, and that when the concentration of HNO3 beyonds the threshold, the strong cut would destroy g-C3N4 seriously and form g-C3N4 ribbons and nanodots.
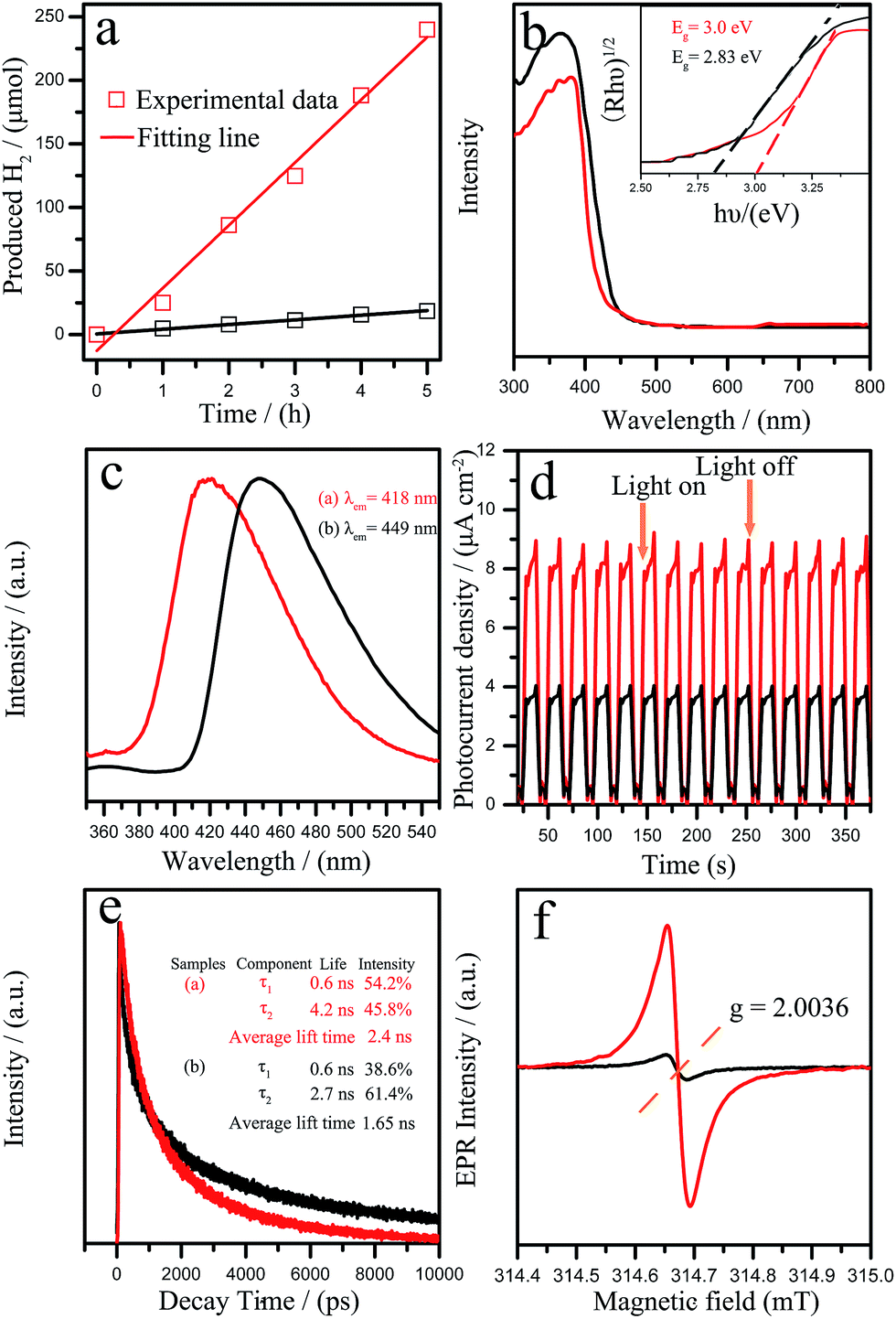
The resultant GCNR was evaluated for photocatalytic H2 production under visible light (λ > 420 nm). As shown in Fig. 3a, it was found that the photocatalytic activity of GCNR showed a significant improvement over the PCN. The H2 evolution rate of GCNR reached 49.3 μmol h−1 was about 19.7 times that of the PCN (2.5 μmol h−1). The apparent quantum yield (AQY) for H2 evolution was also calculated for the GCNR photocatalysts.27 The AQY values for GCNR at 420 nm is estimated as 2.4%. Table 2 summarizes the photocatalytic water splitting based on C3N4 with different morphologies.
 | ||
| Fig. 3 (a) Photocatalytic H2 evolution performance. (b) UV-visible absorption and Tauc plot inset. (c) PL spectra. (d) Transient photocurrent response. (e) Time-resolved PL spectra. (f) EPR spectra of the pristine g-C3N4 (black curve) and GCNR (red curve). | ||
| Morphology (C3N4) | Mass of catalyst | Reactant solution | Lightsource wavelength | Activity (μmol h−1) | Quantum efficiency (%) | Reference |
|---|---|---|---|---|---|---|
| Nanosheet | 20 mg + 3 wt% Pt | 20 mL of 10 vol% triethanolamine aqueous solution | 500 W Xe lamp (>420 nm) | 230 | — | 21 |
| Nanosheet | 50 mg + 3 wt% Pt | 100 mL of 10 vol% triethanolamine aqueous solution | 300 W Xe lamp (>420 nm) | 93 | 3.75 (429 nm) | 13a |
| Macroscopic monolith | 10 mg + 3 wt% Pt | 30 mL of 10 vol% triethanolamine aqueous solution | 300 W Xe lamp (>420 nm) | 29 | — | 14 |
| Nanosheet | 50 mg + 6 wt% Pt | 300 mL of 10 vol% triethanolamine aqueous solution | 300 W Xe lamp (>400 nm) | 33 | — | 22 |
| Holey nanosheet | 10 mg + 3 wt% Pt | 30 mL of 10 vol% triethanolamine aqueous solution | 300 W Xe lamp (>420 nm) | 82.9 | — | 23 |
| Mesoporous hexagonal framework | 20 mg + 3 wt% Pt | 100 mL of 10 vol% triethanolamine aqueous solution | 300 W Xe lamp (>420 nm) | 243 | 6.77 (450 nm) | 24 |
| Holey nanosheet | 50 mg + 3 wt% Pt | 210 mL of 14 vol% triethanolamine aqueous solution | 300 W Xe lamp (>400 nm) | 316.7 | — | 10 |
| This work | 100 mg + 3 wt% Pt | 80 mL of 10 vol% triethanolamine aqueous solution | 500 W Hg lamp (>420 nm) | 49.7 | 2.4 (420 nm) | — |
The excellent photocatalytic hydrogen evolution of GCNR can be attributed to the unique morphology and structures. On one hand, the ultrathin nanostructure of GCNR might provide more active sites during the photocatalytic reaction. Then, the existence of more carbon vacancies of GCNR would contribute to remarkably optical absorption properties and abundant active sites. The optical properties of the samples were studied by UV-vis absorption spectra (Fig. 3b). The derived electronic band gaps from the Tauc plots were 2.83 eV for GCNR and 3.0 eV for pristine sample (inset in Fig. 3b).3a The photoluminescence (PL) (shown in Fig. 3c) further confirmed the above result. Two broad emission peaks at 418 nm and 449 nm were observed for GCNR and PCN, respectively. The blue shift of PL spectra results from the strong quantum confinement effect, which are caused by the decrease of the conjugation length and opposite directions shift of conduction and valence bands.14,28 The photocurrent responses of the samples with typical on–off cycles under visible light irradiation were further investigated shown in Fig. 3d. The photocurrent density of GCNR is 2.1 times larger than that of the pristine catalyst. The results indicated that the separation rate of the photoexcited charge carriers was promoted. Fig. 3e showed the time-resolved fluorescence decay spectra which indicated the average radiative lifetime. The τ2 lifetime component improved from 2.7 ns for pristine g-C3N4 to 4.2 ns for GCNR. The carbon vacancies may be beneficial for hindering the recombination of electrons and holes when they provide trapping sites for photoinduced holes because the improvement of photocurrent and lifetime of charge carriers. Specifically, the average PL lifetime of GCNR (2.4 ns) was 1.45 times longer than that of pristine g-C3N4 (1.65 ns) which was beneficial to improve the transport rate of the photo-generated carriers. This result demonstrated that the separation of electron–hole pairs along the in-plane direction in GCNR nanostructure could be improved due to the anisotropic structure.29 As shown in Fig. 3f, the EPR spectrum of pristine g-C3N4 shows a weak similar response at g = 2.0036. Note the GCNR displays a strong symmetrical signal centering indicates the generation of carbon-based radicals and much higher concentration of unpaired electrons which play an important role in the photocatalytic reaction.14,15 At the same time, the ferromagnetism is generated on the zigzag edges created by the carbon vacancies in the broken honeycomb lattice.23,30 These unsaturated N atoms could act as the paramagnetic centers and attract photoelectrons from the conduction band of GCNR.31
In conclusion, g-C3N4 nanoribbon (GCNR) containing a large number of in-plane carbon vacancies was prepared by controlling the HNO3/H2SO4 volume ratio. Photoluminescence and time-resolved fluorescence decay spectra demonstrate GCNR improves the separation of electron–hole and charge lifetime. Compared to the pristine g-C3N4, the GCNR shows the hydrogen evolution rates improved from 2.5 μmol h−1 to 49.3 μmol h−1 under visible light as a result of the special structure, abundant carbon vacancies and excellent ability of separation of photoexcited electrons and holes. This work provides a practical method to control the structure and photocatalytic activity of g-C3N4, and thus enhance the application of this metal-free, stable and available photocatalyst.
Acknowledgements
This work was supported by NSFC (51572173), STCSM (16JC1402200, 15520720300).Notes and references
- J. Zhang, Y. Chen and X. Wang, Energy Environ. Sci., 2015, 8, 3092 CAS.
- X. Dong and F. Cheng, J. Mater. Chem. A, 2015, 3, 23642 CAS.
- (a) J. Liu, Y. Liu, N. Y. Liu, Y. Z. Han, X. Zhang, H. Huang, Y. Lifshitz, S. T. Lee, J. Zhong and Z. Kang, Science, 347, 970 Search PubMed; (b) X. D Zhang, X. Xie, H. Wang, J. J Zhang, B. C Pan and Y. Xie, J. Am. Chem. Soc., 2013, 135, 18 CrossRef PubMed; (c) X. Zhang, H. Wang, H. Wang, Q. Zhang, J. Xie, Y. Tian, J. Wang and Y. Xie, Adv. Mater., 2014, 26, 4438 CrossRef CAS PubMed.
- (a) S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150 CrossRef CAS PubMed; (b) Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717 RSC; (c) Y. Wang, X. Wang and M. Antonietti, Angew. Chem., Int. Ed., 2012, 51, 68 CrossRef CAS PubMed.
- (a) Y. Zhang, A. Thomas, M. Antonietti and X. C. Wang, J. Am. Chem. Soc., 2009, 131, 50 CrossRef CAS PubMed; (b) S. Yan, Z. Li and Z. Zou, Langmuir, 2009, 25, 10397 CrossRef CAS PubMed.
- X. Jin, V. V. Balasubramanian, S. T. Selvan, D. P. Sawant, M. A. Chari, G. Q. Lu and A. Vinu, Angew. Chem., 2009, 121, 8024 CrossRef.
- Z. Xing, A. M. Asiri, A. O. Al-Youbi and X. Sun, Nanoscale, 2013, 5, 8921 RSC.
- X. Li, X. Wang and M. Antonietti, Chem. Sci., 2012, 3, 2170 RSC.
- D. Zheng, C. Huang and X. Wang, Nanoscale, 2015, 7, 465 RSC.
- P. Yang, J. Zhao, W. Qiao, L. Lia and Z. Zhu, Nanoscale, 2015, 7, 18887 RSC.
- J. Zhang, M. Zhang, C. Yang and X. Wang, Adv. Mater., 2014, 26, 4121 CrossRef CAS PubMed.
- (a) F. Cheng, J. Yan, C. Zhou, B. Chen, P. Li, Z. Chen and X. Dong, J. Colloid Interface Sci., 2016, 468, 103 CrossRef CAS PubMed; (b) J. Yan, C. Zhou, P. Li, B. Chen, S. Zhang, X. Dong, F. Xi and J. Liu, Colloids Surf., A, 2016, 508, 257 CrossRef CAS.
- (a) S. Yang, Y. Gong, J. Zhang, L. Zhan, L. Ma, Z. Fang, R. Vajtai, X. Wang and P. M. Ajayan, Adv. Mater., 2013, 25, 2452 CrossRef CAS PubMed; (b) Y. Yuan, W. Xu, L. Yin, S. Cao, Y. Liao, Y. Tang and C. Xue, Int. J. Hydrogen Energy, 2013, 38, 13159 CrossRef CAS; (c) J. Xu, L. Zhang, R. Shi and Y. Zhu, J. Mater. Chem. A, 2013, 1, 14766 RSC.
- Q. Liang, Z. Li, Z. Huang, F. Kang and Q. Yang, Adv. Funct. Mater., 2015, 25, 6885 CrossRef CAS.
- Z. H. Hong, B. Shen, Y. l. Chen, B. Z. Lin and B. F. Gao, J. Mater. Chem. A, 2013, 1, 11754 CAS.
- (a) M. J. Bojdys, N. Severin, J. P. Rabe, A. I. Cooper, A. Thomas and M. Antonietti, Macromol. Rapid Commun., 2013, 34, 850 CrossRef CAS PubMed; (b) X. She, H. Xu, Y. Xu, J. Yan, J. Xia, L. Xu, Y. Song, Y. Jiang, Q. Zhang and H. Li, J. Mater. Chem. A, 2014, 2, 2563 RSC.
- (a) H. Li, B. Sun, L. Sui, D. Qian and M. Chen, Phys. Chem. Chem. Phys., 2015, 17, 3309 RSC; (b) J. Sun, Y. Deng, J. Li, G. Wang, P. He, S. Tian, X. Bu, Z. Di, S. Yang, G. Ding and X. Xie, ACS Appl. Mater. Interfaces, 2016, 8, 10226 CrossRef CAS PubMed.
- (a) X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. A. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed; (b) L. Tian, S. Yang, Y. Yang, J. Li, Y. Deng, S. Tian, P. He, G. Ding, X. Xie and Z. Wang, RSC Adv., 2016, 6, 82648 RSC.
- (a) G. Zhang, J. Zhang, M. Zhang and X. Wang, J. Mater. Chem., 2012, 22, 8083 RSC; (b) S. Yang, J. Sun, X. Li, W. Zhou, Z. Wang, P. He, G. Ding, X. Xie, Z. Kang and M. Jiang, J. Mater. Chem. A, 2014, 2, 8660 RSC; (c) S. Yang, J. Sun, P. He, X. Deng, Z. Wang, C. Hu, G. Ding and X. Xie, Chem. Mater., 2015, 27, 2004 CrossRef CAS.
- (a) J. Feng, T. Chen, S. Liu, Q. Zhou, Y. Ren, Y. Lv and Z. Fan, J. Colloid Interface Sci., 2016, 479, 1 CrossRef CAS PubMed; (b) S. Yang, C. Zhu, J. Sun, P. He, N. i Yuan, J. Ding, G. Ding and X. Xie, RSC Adv., 2015, 5, 33347 RSC; (c) X. Deng, J. Sun, S. Yang, H. Shen, W. Zhou, J. Lu, G. Ding and Z. Wang, Appl. Phys. Lett., 2015, 107, 241905 CrossRef.
- J. Xu, L. Zhang, R. Shi and Y. Zhu, J. Mater. Chem. A, 2013, 1, 14766 CAS.
- P. Niu, L. Zhang, G. Liu and H. Cheng, Adv. Funct. Mater., 2012, 22, 4763 CrossRef CAS.
- Q. Liang, Z. Li, Z. Huang, F. Kang and Q. Yang, Adv. Funct. Mater., 2015, 25, 6885 CrossRef CAS.
- J. Zhang, F. Guo and X. Wang, Adv. Funct. Mater., 2013, 23, 3008 CrossRef CAS.
- F. Zhao, H. Cheng, Y. Hu, L. Song, Z. Zhang, L. Jiang and L. Qu, Sci. Rep., 2014, 4, 5882 CrossRef CAS PubMed.
- (a) X. Bai, S. Yan, J. Wang, L. Wang, W. Jiang, S. Wu, C. Sun and Y. Zhu, J. Mater. Chem. A, 2014, 2, 17521 RSC; (b) M. T. Schumperili, C. Hammond and I. Hermans, Phys. Chem. Chem. Phys., 2012, 14, 11002 RSC.
- L. Yuan, C. Han, M. Yang and Y. Xu, Int. Rev. Phys. Chem., 2016, 35, 1 CrossRef CAS.
- A. P. Alivisatos, Science, 1996, 271, 933 CAS.
- (a) T. Ma, S. Dai, M. Jaroniec and S. Qiao, Angew. Chem., Int. Ed., 2014, 53, 7281 CrossRef CAS PubMed; (b) Y. Tang, P. Wang, J. Yun, R. Amal and Y. H. Ng, J. Mater. Chem. A, 2015, 3, 15876 RSC.
- S. Sun, J. Magn. Magn. Mater., 2013, 344, 39 CrossRef CAS.
- S. Li, G. H. Dong, R. Hailili, L. Yang, Y. Li, F. Wang, Y. Zeng and C. Wang, Appl. Catal., B, 2016, 190, 26 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra23218c |
| This journal is © The Royal Society of Chemistry 2016 |