
Promotional effects of rare earth elements (Sc, Y, Ce, and Pr) on NiMgAl catalysts for dry reforming of methane†
Yang Cao,
Hongrui Li,
Jianping Zhang,
Liyi Shi and
Dengsong Zhang *
*
Department of Chemistry, Research Center of Nano Science and Technology, Shanghai University, Shanghai 200444, China. E-mail: dszhang@shu.edu.cn; Tel: +86-21-66137152
First published on 16th November 2016
Abstract
In this work, the promotional effects of rare earth elements (Sc, Y, Ce, and Pr) on NiMgAl catalysts derived from layered double hydroxides were investigated for dry reforming of methane (DRM). It was found that the modified catalysts showed improved catalytic stability and coke resistance, which may result from the following favorable properties: enhanced surface basicity, abundant oxygen vacancies, superior redox properties and highly dispersed Ni particles. Notably, the Ce or Pr modified catalysts showed higher performance than the others. It may be that the addition of Ce or Pr can increase the amount of strong basic sites. Furthermore, the coexistence of redox pairs, like Ce3+/Ce4+ or Pr3+/Pr4+, can contribute to the enhancement of redox properties and formation of oxygen vacancies simultaneously. The high oxygen vacancies and excellent redox properties can significantly help to improve the catalytic stability. In situ diffuse reflectance infrared transform spectroscopy analysis illustrated the details of carbonate species forming during the adsorption of CO2, and the transient studies investigated the dynamic changes of the adsorbed intermediate species during catalysis processes for getting deep insight into the catalytic mechanism. Adding various rare earth elements can lead to different chemical environments and electronic effects of the surface, and it is crucial to investigate how these additives affect the behavior of the catalysts surface and interface, thus, identifying the catalytic mechanism via the elucidation of the promotional effects of these properties on the catalytic performance.
1. Introduction
With energy shortages and environmental pollution becoming a serious global problem, syngas emerged as a clean source and has received much attention in the past few decades.1–3 Syngas, primarily composed of CO and H2, is used as a feedstock for Fischer–Tropsch reaction.4,5 Typically, the syngas can be obtained from such reforming reactions as partial oxidation of methane, dry reforming of methane and steam reforming of methane, among which dry reforming of methane (DRM) can utilize the two greenhouse gases (CH4 and CO2) well.2,6,7 The DRM reaction not only reduces the emissions of CH4 and CO2 but also provides a promising way to utilize the natural gas resources.8 Now, the Ni-based catalyst has been extensively used for DRM reaction due to their extensive availability, high activity and low cost.9,10 However, the main problem of Ni-based catalysts is the deactivation of their catalytic performance owning to the loss of active surface by metal sintering and coke deposition.6,11–15 Therefore, the DRM catalysts should be catalytically and mechanically stable under the high temperature conditions. It's still a great challenge to develop highly efficient Ni-based catalysts for DRM reaction.In order to overcome the drawbacks, great efforts have been made to develop coking and sintering resistant Ni-based catalysts. For example, immobilizing Ni nanoparticles in porous shells can inhibit the agglomeration of Ni particles.16 Besides, the catalysts with certain structures like pyrochlore, spinel, and solid solutions showed strong interaction between highly dispersed Ni particles and support, which obviously improved the sintering resistance.17,18 In recent years, layered double hydroxides (LDH) as the alternative 2D materials has gained great interest due to their unique physicochemical properties, such as high dispersed metal species, diversity of composition, and functionality of the structure.19 Zhan et al. reported the Pt-doped Ni-based catalysts derived from the LDH exhibited durable stability during steam reforming of methane.20 In our previous study, we have prepared monolithic Ni-based catalysts derived from LDH to inhibit the Ni species sintering and coking.21,22 Additionally, the rare earth elements as the promoter have been exploited for DRM catalysts.15,23 For instance, the Ce-doped Ni/SBA-16 catalysts can suppress the growth of Ni species,24 and the addition of La can improve the catalytic activity by the enhanced CO2 adsorption.25 Recently, the rare earth elements were employed to promote the performance of catalysts derived from LDH. By modifying the LDH precursor with Y or La, the catalysts showed high activity and selectivity in the catalytic reaction due to the enhanced basicity.26–28 However, aside from the beneficial properties of the rare earth elements, it is essential to understand the different chemical environment and electronic effects of adding various rare earth elements in order to clarify the reaction mechanism through which rare earth elements influence the catalytic performance.13,29
Herein, the study aims to clarify the promoting effect of various rare earth elements (Sc, Y, Ce, and Pr) on the catalytic performance of NiMgAl catalyst derived from LDH during DRM reaction. Based on the characterization results of the catalysts, the detailed study provide valuable insight for how the doped rare earth elements influence the catalysts surface basic sites, oxygen vacancies, redox properties and dispersion of Ni particles, and the these favorable properties can result in different effects on the nature of the catalysts as well as the catalytic activity and stability, thus, the reaction mechanism can be clarified from the deep investigation.
2. Experimental
2.1. Reagents and materials
The rare earth nitrates were purchased from Diyang Chemical Company Ltd, and the other chemicals were purchased from Sinopharm Chemical Reagent Company China. All the chemicals were used without any further purification. Deionized water was used for all applications.2.2. Catalyst preparation
The catalysts containing various rare earth elements (RE: Sc, Y, Ce, and Pr) derived from LDH precursor were prepared by co-precipitation method and activated by the calcination and reduction. In the synthesis of LDH precursor, 0.15 mol of NaOH aqueous solution was quickly added into mixed salt solution containing amount of metal nitrates (Ni![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Mg:Al:RE in a 0.86:5.14:1.85:0.15 mmol ratio) under vigorous stirring. After being stirred 10–15 min, washed the sol-like samples twice with deionized water and gained by centrifugation, then dispersed into deionized water and transferred into Teflon autoclave, the solution were heated at 120 °C for 5 h. Finally, the pale green powders were obtained by reduced pressure distillation through a rotary evaporator. The samples were denoted as NiMgAl(RE)-LDH, where rare earth elements represent Sc, Y, Ce, Pr elements respectively. The as-prepared NiMgAl(RE)-LDH samples were calcined at 550 °C for 6 h under air atmosphere denoted as NiOMgAl(RE), and reduced under the hydrogen at 900 °C, the resulting catalysts denoted as NiMgAl(RE).
:Mg:Al:RE in a 0.86:5.14:1.85:0.15 mmol ratio) under vigorous stirring. After being stirred 10–15 min, washed the sol-like samples twice with deionized water and gained by centrifugation, then dispersed into deionized water and transferred into Teflon autoclave, the solution were heated at 120 °C for 5 h. Finally, the pale green powders were obtained by reduced pressure distillation through a rotary evaporator. The samples were denoted as NiMgAl(RE)-LDH, where rare earth elements represent Sc, Y, Ce, Pr elements respectively. The as-prepared NiMgAl(RE)-LDH samples were calcined at 550 °C for 6 h under air atmosphere denoted as NiOMgAl(RE), and reduced under the hydrogen at 900 °C, the resulting catalysts denoted as NiMgAl(RE).
2.3. Catalyst characterization
Elemental chemical analysis was obtained by inductively coupled plasma-atomic emission spectrometer (ICP-AES). Transmission electron microscope (TEM) was performed on JEOL JEM-200CX. Nano Measurer was used to count the Ni particle size distribution, more than 100 particles were measured. The XRD pattern was performed using a Rigaku D/MAX-RB X-ray diffractometer with Cu Kα radiation. X-ray photoelectron spectroscopy (XPS) was performed using a RBD upgraded PHI-5000C ESCA system, and the C 1s (284.6 eV) binding energy was used to calibrate the spectra.The H2-temperature programmed reduction (H2-TPR) measurement was performed to illustrate the reducibility of catalysts. Prior to reduction measurement, the catalysts (80 mg) were purged at 300 °C for 30 min with high purity nitrogen to remove physically adsorbed water, then reduced in 10% H2 of N2 atmosphere (30 mL min−1) at a ramp rate of 10 °C min−1 up to 900 °C.
The CO2-temperature programmed desorption (CO2-TPD) measurement was performed to study the catalysts basicity. Prior to TPD measurement, the catalysts (120 mg) were purged in a high purity He flow at 300 °C for 30 min, then exposed to CO2 (30 mL min−1) for 1 h followed by He purging for 30 min to remove physical absorbed CO2. After purged treatment, the catalysts were heated in a high purity He flow from room temperature to 750 °C at a ramp rate of 10 °C min−1.
The O2-temperature programmed oxidation (O2-TPO) measurement was carried out to illustrate carbon species on the surface. The catalysts (50 mg) were heated in 10% O2 of N2 flow (30 mL min−1), the temperature increased at a rate of 10 °C min−1 until 800 °C.
The thermogravimetric (TG) analysis was performed on NETZSCH STA 449 F1 to illustrate the amount of carbon deposition on the surface. The spent catalysts were heated in 40% O2 of N2 flow from room temperature to 900 °C at a ramp rate of 10 °C min−1.
N2 adsorption–desorption isotherm and hydrogen chemisorption were performed on a Quantachrome instrument. The specific surface areas were obtained using BET equation. The BJH method was used to calculate the pore size distribution. For the hydrogen chemisorption, the catalysts were purged with high purity He and reduced in H2 atmosphere, then the catalysts were purged with He and cooled in vacuum to room temperature for the chemisorption.
In situ diffuse reflectance infrared transform spectroscopy (DRIFTs) analysis was applied to the study of the catalysts behavior under various conditions. In situ DRIFTs was performed on a Nicolet 6700 spectrometer with a Harrick Scientific DRIFT cell and a mercury–cadmium–telluride (MCT) detector. Spectra were obtained by collecting 64 scans or 8 scans at 4 cm−1 resolution in the Kubelka–Munk format. In a typical experiment, the catalysts were purged with N2 (50 mL min−1) for 0.5 h at 300 °C, and then cooled to room temperature to get a background spectrum, and this spectra was subtracted from the catalysts spectra for each measurement. In a typical experiment, the catalysts were exposed to CO2 (45 mL min−1) atmosphere and then heated to the desired temperature. In another experiment designed to illustrate the changes of the species with CH4 introducing, the catalysts were exposed to CO2 (45 mL min−1) for 1 h at 550 °C and then exposed to CH4 (45 mL min−1), holding at this temperature.
2.4. Catalyst test
The catalytic reactions were tested under steady state conditions. Approximately 120 mg catalysts were loaded into quartz tube with thermal conductivity detector. The feed, a mixture of then a mixture of CO2 and CH4 (molar ratio CO2/CH4 = 1) was introduced at a gas hourly space velocity (GHSV) of 15000 mL·(g h)−1, and the temperature heated up to 750 °C for 20 h at ramp rate of 10 °C min−1. The date collection is used by online gas chromatography equipped with thermal conductivity detector. The obtained experimental data were tested more than three times under the same condition. The final figures are the average of the three sets of data. The relative errors are obtained by calculating the difference between the experimental data and the average values.
3. Results and discussion
3.1. Characteristics of fresh catalysts
The XRD patterns of NiMgAl(RE)-LDH were shown in Fig. 1a. The samples exhibited typical reflections of LDH phases, 2θ = 11.7°, 23.5°, 35.1°, 47.3°, and 62.6°.19,30 The diffraction profile confirmed that Ni2+, Mg2+, Al3+and RE3+ had been constructed in the LDH structure. Compared with NiMgAl-LDH, the rare earth elements modified samples showed lower crystallization phases, which may result from the distorted LDH structure. The differences of ionic radii and electronegativities among various rare earth elements may lead to the reconstructed structure.26,27 Moreover, the layer distance is also a significant factor in which the larger ions can insert into the LDH structures.27,31,32 Notably, the NiMgAlCe and NiMgAlPr catalysts possess CeO2 (JCPDS no. 34-0394) and Pr6O11 (JCPDS no. 41-1219) species, respectively. This can be attributed to their higher ionic radii and the lower electronegativities compared with Al3+, which may prevent part of RE3+ inserting into the LDH galleries and these ions formed their corresponding oxides the surface.27 However, Sc3+ or Y3+can be easily introduced into the LDH structure due to their smaller ionic radii during the preparation of LDH precursor. As shown in Fig. 1b, all NiOMgAl(RE) samples showed the diffraction peaks of Mg(Ni, Al, RE)O periclase phase,33 suggesting that the Ni species existed in the periclase phase. Moreover, the CeO2 phase was still observed over the NiMgAlCe sample, there was no significant diffraction peaks of Pr6O11 phase. The Pr element on the catalysts surface was detected by XPS and showed a sharp peak, suggesting the presence of the Pr, and thus, we supposed that the nonexistence of Pr may come from highly dispersive Pr6O11 phase on the surface. On the other hand, the nonexistence of Pr might come from the lower quantity of Pr6O11 on the surface. Fig. 1c showed the XRD patterns of NiMgAl(RE) catalysts. All catalysts maintained a periclase phase up to 900 °C. Meanwhile, the typical diffraction peak of Ni phase was observed, indicating the Ni2+ can be reduced under the reduction condition. | ||
| Fig. 1 XRD patterns of (a) NiMgAl(RE)-LDH, (b) NiOMgAl(RE), and (c) NiMgAl(RE) samples. | ||
The textural properties of NiOMgAl(RE) samples and NiMgAl(RE) catalysts were shown in Fig. 2. A summary of average pore diameter and specific surface area were shown in Table 1. The isotherm of NiOMgAl(RE) samples exhibited type IV hysteresis, representing a typical mesostructured feature with the BET specific surface area of 252 m2 g−1, 245 m2 g−1 (Sc), 236 m2 g−1 (Y), 232 m2 g−1 (Ce), 245 m2 g−1 (Pr), respectively. Due to the structure restructured by introducing rare earth elements, which may result in the decrease of the specific surface area. As shown in Fig. 2b, the narrow pore diameter distribution of NiOMgAl(RE) sample exhibited two peaks at 1.7 nm corresponded to the main pores and at 6.0–7.5 nm corresponded to the large pores. After the reduction, the NiMgAl(RE) catalysts exhibited a significant decrease in surface areas and a remarkable capillary condensation in the mesoporous range from 7.8 nm to 9.6 nm presented in Fig. 2d. The specific surface area and the pore size of NiMgAl(RE) catalysts showed a distinct change. The specific surface area decreased, and the pore size of reduced catalysts increased. It could be explained by the appearance of restructured pore walls. After reduction treatment, the small pores can be transformed into large pores. Meanwhile, the lower specific surface area could be ascribed to the blocked pores which may be due to the reduced Ni particles loaded into the pore channels.
 | ||
| Fig. 2 N2 adsorption–desorption isotherm of (a) NiOMgAl(RE) samples and (c) NiMgAl(RE) catalysts samples; pore size distributions of (b) NiOMgAl(RE) samples and (d) NiMgAl(RE) catalysts. | ||
| Catalyst | Calcined catalysts | Reduced catalysts | ||
|---|---|---|---|---|
| BET surface area (m2 g−1) | Pore diameter (nm) | BET surface area (m2 g−1) | Pore diameter (nm) | |
| NiMgAl | 252 | 1.7 | 109 | 9.6 |
| NiMgAlSc | 245 | 1.7 | 125 | 7.8 |
| NiMgAlY | 236 | 1.7 | 126 | 9.6 |
| NiMgAlCe | 232 | 1.7 | 107 | 7.8 |
| NiMgAlPr | 245 | 1.6 | 113 | 7.8 |
Fig. 3 showed the TEM images and HRTEM images of the NiMgAl(RE) catalysts. It was found that the Ni particles were nanosized and homogenously dispersed in the layered structure. The particles size distribution of NiMgAl(RE) catalysts showed that the Ni particles had diameters in a 10–15 nm range. Nevertheless, the addition of rare earth elements did not increase in the size of the Ni particle significantly. It can be assumed that the confinement effect of LDH precursor can be able to achieve highly dispersed Ni particles and the doped rare earth elements can stabilize the structure of NiMgAl(RE) catalysts.21,34
 | ||
| Fig. 3 TEM, HRTEM images and particle size distribution of (a) NiMgAl, (b) NiMgAlSc, (c) NiMgAlY, (d) NiMgAlCe, and (e) NiMgAlPr catalysts (TEM scale bar: 100 nm, HRTEM scale bar: 10 nm). | ||
The H2-TPR was performed to illustrated the redox properties of NiMgAl(RE) catalysts.35 As shown in Fig. 4a, it was found that all NiMgAl(RE) catalysts showed a single asymmetric peak indicated the reduction of Ni2+ to Ni0.36 Additionally, all reduction peaks could be observed above 750 °C which may attribute to the formation of Mg(Ni, Al, RE)O periclase, suggesting the stronger interaction between NiO and support.37 The higher reduction temperature may attribute to the strong interaction between Ni and support. Typically, the strong metal–support interaction can prevent the migration of Ni species effectively, which benefit to the inhibition of metal sintering. For the NiMgAl catalyst, the highest reduction peak at approximately 835 °C, while the highest reduction peaks of Sc, Y, Ce, and Pr modified catalysts were at 799 °C, 790 °C, 766 °C and 775 °C, respectively. The reduction peak of rare earth elements modified catalysts shifted to the lower temperature. This phenomenon can be confirmed by that the reduction of NiO can be controlled by the electron transfer capability and the oxygen storage capacity.13,29 It has been demonstrated that the coexistence of redox pairs, like Ce3+/Ce4+or Pr3+/Pr4+, can facilitate the electron transport, meanwhile the redox property of rare earth elements is one of the important properties for the formation of oxygen vacancies.8,23,38 The existence of CeO2 and Pr6O11 can change their oxidation state easily from 4+ to 3+, therefore, the formation of oxygen vacancies can be facilitated (Fig. S2†).29,39–41 The increased oxygen vacancies and fast electron transfer capability can promote the reduction of Ni2+, and thus, the reduction peak shift to the lower temperature (Fig. S1†). In addition, the high dispersion of NiO specie also contributes to the reduction of Ni2+.24,38 Since the catalysts doped with various rare earth elements, the catalysts showed different interaction between Ni and support, which can improve the dispersion of NiO specie on the surface and benefit to the reduction of Ni2+.42
 | ||
| Fig. 4 H2-TPR profiles (a), Ni 2p XPS patterns (b), O 1s XPS patterns (c) and CO2-TPD profiles (d) of NiMgAl(RE) catalysts. | ||
The XPS analysis was performed to determine the chemical environment and oxidation states. XPS whole spectra of NiMgAl(RE) catalysts showed sharp peaks of Ni, Mg, Al, RE and O (Fig. S3†). Fig. 4b and c showed Ni 2p and O 1s XPS patterns of NiMgAl(RE) catalysts, and the related parameters were listed in Table 2. Typically, Ni 2p binding energy generally used to determine the chemical environments and oxidation states of Ni species during DRM reaction. The NiMgAl(RE) catalysts gave the typical state of Ni2+ with two spin orbit components of Ni 2p1/2 and Ni 2p3/2. It could be observed for all catalysts that Ni 2p1/2 binding energy ranged at 873.6–874.0 eV, accompanied by satellites around 880.4–882.8 eV. It is noted that the rare earth oxides might act as an electron donor which could transfer partial electrons to metallic Ni, leading to an increase in the d-electron density of the surface Ni species, and thus, the binding energy of Ni 2p1/2 shifted to the lower value.25 Among the catalysts, the Ni 2p1/2 binding energy of Ce or Pr modified catalysts shifted to the lower value, suggesting that the addition of Ce or Pr can promote the electron transfer, and thus, part of Ni2+ can be easily reduced to Ni, which is consistent with the results of H2-TPR. The phenomena of electron transfer between Ni species and other metals has been reported previously. For example, S. A. Nikolaev et al. reported the existence of the electron transfer from gold to oxidized nickel, as well as in other catalysts such as Sr-doped Ni-La2O3 catalysts and Mo-NiO/Al2O3.43–45 It could be seen from Table 2 that the addition of Sc or Y did not affect the binding energy of the surface Ni species. In addition, all NiMgAl(RE) catalysts showed a Ni 2p3/2 main peak at 855.6–856.2 eV accompanied by satellites around 862.0–862.6 eV. This indicated that part of Ni2+ can be reduced to Ni0, while part of them remained as Ni2+ in the periclase (Mg(NiAlRE)O) species. Moreover, the Ni 2p3/2 could illustrate the interaction between Ni2+ species and support. Different strength of the chemical bond formed on the surface might result in different Ni species and support interactions. The pure NiO binding energy of Ni 2p3/2 was 854.5 eV, and the higher binding energy of NiMgAl(RE) catalysts suggested a strong interaction between Ni and support.42
| Catalyst | Binding energy and concentration of Oα | Basic sites distribution and relative contents | |||||
|---|---|---|---|---|---|---|---|
| Ni 2p3/2 (eV) | Ni 2p1/2 (eV) | Oα/O (%) | Weak basic sites | Moderate basic sites | Strong basic sites | Total | |
| NiMgAl | 856.2 | 874.0 | 65.96 | 1.00 | 2.21 | 2.33 | 5.54 |
| NiMgAlSc | 856.2 | 874.0 | 69.92 | 1.37 | 2.39 | 2.57 | 6.33 |
| NiMgAlY | 856.2 | 874.0 | 71.62 | 1.84 | 3.00 | 3.06 | 7.91 |
| NiMgAlCe | 855.6 | 873.6 | 73.69 | 1.77 | 3.10 | 3.83 | 8.70 |
| NiMgAlPr | 856.0 | 873.9 | 73.46 | 1.57 | 2.66 | 3.81 | 8.04 |
The XPS spectra of O 1s for NiMgAl(RE) catalysts were fitted into two peaks. The low binding energy was assumed to the lattice oxygen (denoted as Oβ), while the high binding energy was the adsorbed oxygen species onto the surface (denoted as Oα) which was identified as the adsorbed O2, OH− and CO32−.38,46 A summary of O 1s spectra were presented in Table 2, it showed that various rare earth elements could improve the ratio of Oα/O compared with unmodified catalyst. Among the catalysts, the relative percentage of absorbed oxygen could be found to increase in the following order: unmodified catalysts (65.96%), Sc (69.92%), Y (71.62%), Pr (73.46%), and Ce (73.69%). Generally, the absorbed oxygen is much more reactive than the lattice oxygen due to its high mobility, therefore, the presence of active oxygen species on the surface played an important role in suppressing the carbon deposition.29 Since deposited carbon can react with the surface absorbed oxygen to form easy removable CO2 gases, which may contribute to preventing catalyst deactivation during DRM reaction. In addition, Ce or Pr modified catalysts possess more oxygen vacancies, which allow more oxygen to be absorbed on the surface to participate in the oxidation of deposited carbon.38
As well known, LDH had been used to attract CO2 adsorbents.47,48 The NiMgAl(RE) catalysts derived from LDH were two-dimensional nanostructured materials, which were mainly composed of Al2O3 and MgO species. Moreover, the presence of MgO could generate stronger basic sites through the increased activity for CO2 adsorption.49 The catalysts basicity has a significant influence on the catalytic activity and stability. Fig. 4d showed the CO2-TPD profiles of NiMgAl(RE) catalysts, which illustrated the promoting effect of various rare earth elements as the promoter on the adsorption of CO2. The desorption temperature can illustrate the strength of basic sties and the areas of CO2 desorption peak can demonstrate the amount of basic sites. It was found that all catalysts showed three types of peaks around at 150 °C, 220 °C and 400 °C, and these peaks were ascribed to the weak basic sites, moderate basic sites and strong basic sites, respectively. The weak and moderate basic sites can be related to the surface OH− and Lewis acid–base pairings, and the strong basic sites can be correlated with the surface O2−.2,40 Table 2 demonstrated the basic sites distribution and the relative contents. The results showed that the addition of rare earth elements can promote the adsorption of CO2 effectively. The total basicity of Y modified catalysts is greater than Sc doped catalyst, it is probably due to that the electronegativity of Y is lower.26,27 Among the rare earth elements modified catalysts, the Ce or Pr modified catalysts showed the much stronger adsorption of CO2, especially the amount of strong basic sites increased significantly. It may be due to the coexistence of the redox pairs, like Ce3+/Ce4+ and Pr3+/Pr4+, which can facilitate the electron transfer and promote the formation of oxygen vacancies to provide more active sites for the absorption of CO2.8,38 The presence of rare earth elements led to the increased basicity, which can reflect by the exceptional long-term stability. Generally, the catalysts are often affected by carbon accumulation on the catalysts surface during DRM reaction.9,50 There are two general reactions which could result in carbon deposition: CO disproportionation (2CO = C + CO2) and CH4 decomposition (CH4 = C + 2H2).24,30 While, the deposited carbon can be gasified by CO2 (C + CO2 = 2CO), thereby enhancing the adsorption of CO2 can effectively decrease the carbon deposition and prevent the catalyst deactivation.
3.2. Catalytic performances for DRM reaction
The effect of various rare earth elements on the catalytic performance over NiMgAl catalysts during DRM reaction were shown in Fig. 5. It was found that all NiMgAl(RE) catalysts showed the high initial CH4 and CO2 conversions which may result from the high dispersion of Ni species (Table S2†). As NiMgAl(RE) catalysts were prepared by the calcination and reduction of NiMgAl(RE)-LDH, the LDH precursor can contribute to the well dispersed and thermally stable Ni species.21,30,33 However, with the DRM reaction going on, the Ni particles could agglomerate or be covered by carbon deposition, resulting in the catalysts deactivation.51,52 Furthermore, the thermodynamic equilibrium has been calculated which consists of DRM and RWGS reactions (Equilibrium Composition Block of HSC software, version 6.0). Under the reaction condition, the CH4 conversions over NiMgAl(RE) catalysts were slightly lower than the thermodynamic equilibrium conversion, illustrating the existence of side reactions (Fig. S5†). For the NiMgAl catalyst, the CH4 conversion decreased by 5.74% after 20 h on stream as presented in Fig. 5a. By contrast, the catalysts modified by Sc, Y, Ce or Pr showed a drop in CH4 conversion by 3.57%, 2.93%, 1.22% and 1.61%, respectively. Additionally, the catalysts containing various amounts of rare earth elements showed the different catalytic stability (Fig. S4†). It can be noted that the addition of rare earth elements can enhance the catalytic stability. Notably, there was no significant activity loss for NiMgAlCe catalyst after a long-term reaction. The difference in the catalytic stability of NiMgAl(RE) catalysts may result from the deposited carbon as well as the sintering of Ni particles.50,53 The obtained results suggest that the addition of rare earth elements can contribute to maintaining their catalytic durability against carbon deposition and metal sintering. | ||
| Fig. 5 Plots of (a) CH4 conversion, (b) CO2 conversion and (c) H2/CO ratio of NiMgAl(RE) catalysts versus time on stream. | ||
As shown in Fig. 5b, the conversion of CO2 over all catalysts were higher than that of CH4, even though CH4 and CO2 fed at the ratio of 1:1. This indicates that there exist reverse water–gas shift (RWGS) reaction.54,55 The DRM reaction (CH4 + CO2 → 2H2 + 2CO ΔH298 K = +247 kJ mol−1) is typically influenced by the simultaneous occurrence of some side reactions: reverse water–gas shift reaction (CO2 + H2 → CO + H2O ΔH298 K = +41 kJ mol−1), CO disproportionation (2CO → C + CO2 ΔH298 K = −172 kJ mol−1) and CH4 decomposition (CH4 → C + 2H2 ΔH298 K = +75 kJ mol−1). Previous work suggest that there is a thermodynamic potential for these reactions with gas mixtures containing H2, CO, CO2, and CH4 above 1000 K.56,57 The CO2 conversion is still constant due to the more serious RWGS reaction. The RWGS reaction is thermodynamically favored at higher temperature. Previous studies have widely demonstrated the existence of RWGS reaction and its faster reaction process. Although the RWGS reaction existed, the CH4 dissociation is well demonstrated as the determining step for the DRM reaction previously.56,57 Therefore, we supposed that the maintained CO2 conversion and the lower H2/CO ratio can be mainly ascribed to the RWGS reaction. In addition, the conversion of CO2 is a little higher than the thermodynamic equilibrium which might be due to the existence of the side reactions. On one hand, the RWGS reaction could consume part of the CO2. On the other hand, the NiMgAlCe catalyst possessed abundant strong basic sites, the absorbed CO2 could form various carbonates which were considered as the active species for the DRM reaction and the removal of deposited carbon.64 It is noted that the CO2 conversion might contribute to the DRM reaction and the side reactions, and the former is considered as the main step. Therefore, the conversion of CO2 is higher than thermodynamic equilibrium. In addition, the carbon formation is the key factor during the DRM reaction. Typically, the carbon deposited on the catalysts surface is mainly due to the CO disproportionation and CH4 decomposition. The CO disproportionation is exothermic, while the CH4 decomposition is endothermic. The thermodynamic data suggest that the CO disproportionation is considered as the main contributor to carbon deposition. Due to the RWGS reaction could consume part of H2, the CO disproportionation could consume part of CO. The coexistence of CO disproportionation and RWGS might lead to the H2/CO maintained. This phenomenon was also observed by Y. Yin groups and R. Lau groups.58,59 Furthermore, the amount of the deposited carbon evaluated by TG and TPO can illustrate the existence of the side reaction. For the NiMgAlCe catalyst, the H2/CO ratio is higher and the amount of deposited carbon is lower than the others, suggesting the addition of Ce can inhibit the side reaction effectively.14
To gain insight into the intrinsic activities of the NiMgAl(RE) catalysts, the TOF (550 °C) normalized by surface Ni atoms from H2 chemisorption were compared, the results were summarized in Table 3. Compared with the NiMgAl catalyst, the rare earth elements modified catalysts showed the enhanced TOF value after 8 h reaction time. The NiMgAl catalysts gave a TOFCH4 value of 2024 h−1, while, the Sc, Y, Ce, and Pr modified catalysts gave the TOFCH4 value of 2226 h−1, 2190 h−1, 2349 h−1 and 2285 h−1, respectively. Notably, the highest TOF value noticed over the NiMgAlCe catalyst is due to the result of high dispersion of Ni nanoparticles, therefore, more active sites can be achieved from higher dispersed Ni particles which is benefit to the enhancement of the catalytic activity and stability.
| Catalyst | Nib (wt%) | Ni dispersionc (%) | Conversion (CH4)d (%) | TOFCH4e (h−1) | |
|---|---|---|---|---|---|
| t = 0 h | t = 8 h | ||||
| a Condition: 50 mg of catalysts, CH4:CO2 = 1:1, 45 mL min−1 per reactor, temperature: 550 °C, time: 8 h.b Determined by ICP.c Determined by H2 chemisorption (Er = ±6.4%).d CH4 conversion at 8 h of the DRM reaction (Er = ±0.70%).e In moleCH4 per h per mole sufNi. |
|||||
| NiMgAl | 16.01 | 9.86 | 7.24 | 16.59 | 2024 |
| NiMgAlSc | 15.98 | 10.03 | 8.43 | 21.20 | 2226 |
| NiMgAlY | 15.62 | 10.86 | 9.25 | 22.37 | 2190 |
| NiMgAlCe | 14.56 | 11.88 | 9.92 | 23.99 | 2349 |
| NiMgAlPr | 15.22 | 11.68 | 9.45 | 23.23 | 2285 |
3.3. Characteristics of spent catalysts
The XRD patterns of spent NiMgAl(RE) catalysts after 20 h of reaction were shown in Fig. 6a. The crystal structure and crystalline phases of the spent NiMgAl(RE) catalysts showed no difference as compared with those of the fresh catalysts except a broad peaks of carbon ranged from 25° to 26°.37,60 The strong intensity of surface carbon over NiMgAl catalysts suggest that plenty of carbon deposited on the surface, which may lead to catalysts deactivation. The similar peaks with lower intensity were also observed on rare earth elements modified catalysts, suggesting minor carbon deposited on the rare earth elements promoted catalysts. | ||
| Fig. 6 XRD patterns (a), O2-TPO profiles (b), and TG profiles (c) of spent NiMgAl(RE) catalysts. | ||
Further insights into the deposited carbon over the spent NiMgAl(RE) catalysts were studied by O2-TPO depicted in Fig. 6b. Three kinds of carbon species were identified during DRM reaction, designated as Cα, Cβ and Cγ.46,61 The active Cα species appeared at around 300 °C, which were ascribed to the amorphous carbon and mainly formed via CH4 decomposition at the early stage of the DRM reaction. The Cβ species were appearing at around 500 °C attributed to graphitic carbon, and the Cγ species were above 600 °C corresponded to the carbon nanotubes. All the spent NiMgAl(RE) catalysts showed three main peaks located at 500 °C (I), 575 °C (II) and 650 °C (III), respectively. According to the results shown in Table 4, the content of carbon deposited over the spent NiMgAl(RE) catalysts followed the order: NiMgAl > NiMgAlSc > NiMg-AlY > NiMgAlPr > NiMgAlCe. The main peaks of NiMgAl catalyst appeared at 575 °C and 650 °C were assigned to the graphitic carbon, suggesting the higher oxidation temperature. Meanwhile, the NiMgAl catalyst showed the largest amount of deposited carbon, and thus, the surface Ni particles may be covered or encapsulated by the graphitic carbon, leading to the catalysts deactivation after long-run tests. While, Sc, Y, Ce, or Pr modified catalysts showed a limited amount of deposited carbon compared with unmodified catalyst. For the NiMgAlCe catalyst, the main peak was located at 500 °C which indicate that the carbon can be easily removed during DRM reaction. Since the Ce modified catalyst showed the lowest amount of deposited carbon, a possible explanation for the decrease in carbon deposition is that the addition of Ce can provide more surface active oxygen species through the fast redox cycling. The carbon deposition is a dynamic reaction process. Consequently, the fast redox cycling can promote the gasification of deposited carbon.2 Furthermore, the rare earth elements modified catalysts showed the much stronger adsorption of CO2, showing a significant contribution in the process of carbon removal.
| Spent catalyst | O2-TPO | TG | |||
|---|---|---|---|---|---|
| Peak I | Peak II | Peak III | Total | Weight loss (%) | |
| NiMgAl | 1.00 | 4.54 | 4.68 | 10.22 | 47.22 |
| NiMgAlSc | 1.33 | 3.69 | 4.68 | 9.47 | 38.14 |
| NiMgAlY | 1.64 | 3.42 | 2.77 | 7.82 | 34.44 |
| NiMgAlCe | 3.14 | 2.32 | 0.42 | 5.88 | 30.92 |
| NiMgAlPr | 3.44 | 2.55 | 1.77 | 7.76 | 33.76 |
Further quantification of deposited carbon was tested by TG presented in Fig. 6c. There was a 2–3 wt% weight loss below 100 °C, which can be assigned to the H2O gasification. Except the loss of adsorbed water, there were 47.22%, 38.14%, 34.44%, 30.92% and 33.76% weight percent carbon on the spent NiMgAl catalyst as shown in Table 4, and the Sc, Y, Ce, Pr modified catalysts, respectively. The highest carbon percent on the spent NiMgAl catalyst agreed with the O2-TPO profiles and the strong signals of carbon observed on the XRD patterns. Based on the TG analyses, the initial step of weight loss ranged 100 °C to 300 °C could correspond to the easily oxidized carbon. While, the graphitic carbon or carbon nanotubes can be oxidized above 500 °C, which are considered as the main reason for the deactivation of all catalysts. An obvious weight loss decrease could be found on the rare earth elements modified catalysts, especially, the Ce and Pr modified catalysts showed the lowest amount of deposited carbon due to their abundant basic sites and oxygen vacancies.
3.4. In situ DRIFTs and transient studies
The adsorption property is the main factors in heterogeneous catalytic reactions. The CO2-TPD could quantify the adsorptive amounts, while, the in situ DRIFTs could provide the further information about the adsorption form of the gas to clarify the details of the reactions occurring during the adsorption of CO2 and CH4. Due to the favorable catalytic stability, the NiMgAlCe catalyst is chosen for further investigation by in situ DRIFTs. Fig. 7a showed the DRIFTs analyses of CO2 desorption over the NiMgAlCe catalyst. The catalyst were pre-adsorbed by pure CO2 at room temperature for 1 h, then the in situ DRIFTs were collected during the desorption process. There were four mainly asymmetric and symmetric stretch of carbonates at the 30 °C, the shoulders at 1226 cm−1, 1421 cm−1, 1549 cm−1 and 1646 cm−1 corresponded to the symmetric stretch of bidentate carbonates, symmetric stretch of monodentate carbonates, asymmetric stretch of monodentate carbonates and asymmetric stretch of bidentate carbonates, respectively (Fig. S6 and Table S1†).18,62,63 Various carbonate species could be observed from the shoulders and weak peaks after the adsorption of CO2, and large amounts of carbonate species could be generated by the reaction with the absorbed CO2.18 Along with the increase in temperature, the intensity of the bands from all the carbonates showed a steady decrease until the peaks totally disappeared at 400 °C, the results are assigned to the partial decomposition of the carbonates. Compared with the peak of bidentate carbonates, it can be noticed that the peak of monodentate carbonates can still be detected at 350 °C, suggesting the CO2 absorbed on the catalysts existed mainly in the form of monodentate carbonates. The intensity of four types of carbonates bands disappeared at 400 °C. While, in the actual DRM reaction, the temperature was above 700 °C, the high reaction temperature resulted in that the catalysts surface exist a fast adsorption–desorption of CO2.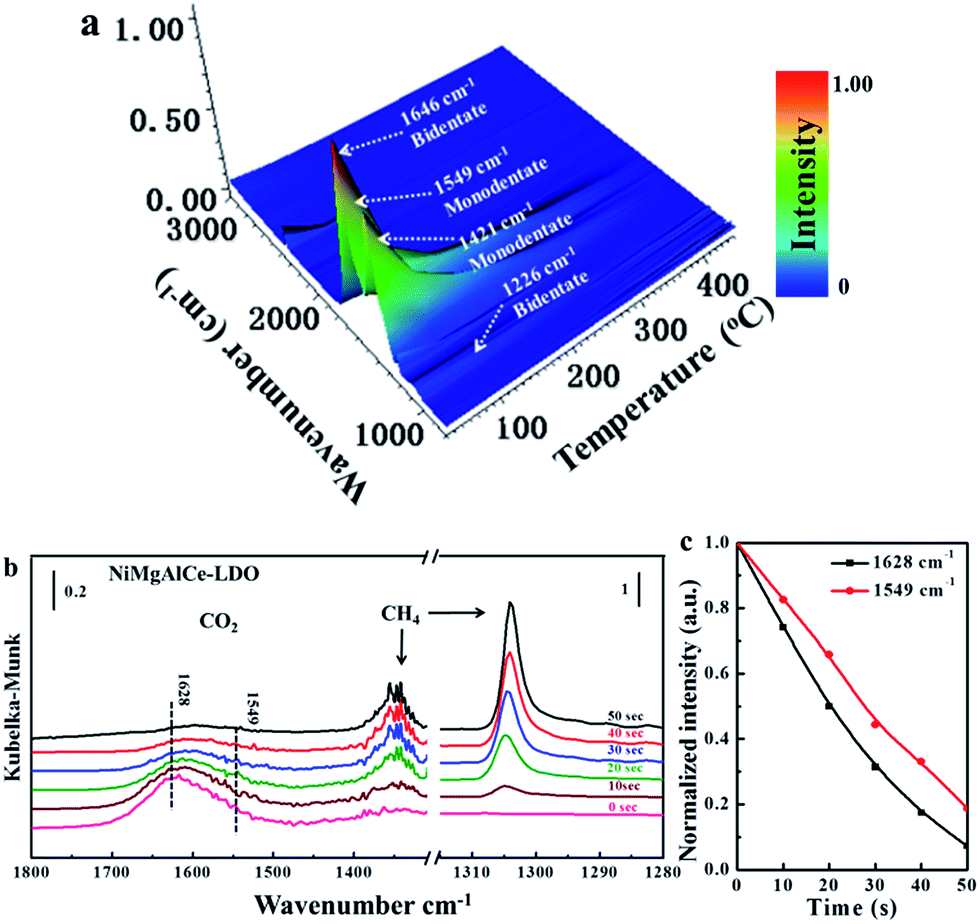 | ||
| Fig. 7 (a) In situ DRIFT spectra of CO2 desorption from NiMgAlCe catalyst as a function of time; (b) in situ DRIFT spectra comparing the changes of CO2 adsorbed into the NiMgAlCe catalyst after the catalysts were exposed to CH4; (c) consumption of different carbonates species at 550 °C upon passing CH4. | ||
Transient reaction studies by in situ DRIFTs were performed to identify adsorbed intermediates species to illustrate the possible reaction process. The DRIFTs of the NiMgAlCe catalyst was shown in Fig. 7b. After the adsorption of CO2, the band at 1628 cm−1 and 1549 cm−1 was attributed to the asymmetric stretch of bidentate carbonates and asymmetric stretch of monodentate carbonates with distinct contributions, pointing to the presence of different carbonate species. However, with the CH4 introducing, the intensity of the bands from 1628 cm−1 and 1549 cm−1 peak decreased significantly, while 1342 cm−1 and 1310 cm−1 peak assigned to the characteristic absorption peak of CH4 increased significantly. Generally, the CO2 could be adsorbed on the surface and formed respective carbonates. To compare the reactivity of two carbonate species, the different species at 550 °C was recorded as a function of time. The bidentate carbonates and monodentate carbonates decreased with CH4 introducing which indicate that CH4 react with the carbonates on the surface leading to the decrease of carbonates. The reactivity of carbonates on NiMgAlCe catalyst showed that the reaction of bidentate carbonates with the CH4 introducing were relatively rapid, which suggested the monodentate carbonates were more favorable for CH4 conversion. Additionally, the formed carbonates can remove carbon from Ni particles, which is assumed to inhibit coking, explaining the enhanced catalytic stability of the promoted catalysts.64
3.5. Possible catalytic mechanism
The rare earth elements as the promoter played an important role in the development of highly efficient DRM catalysts. All the results demonstrate the promoting effects of the additives on the surface basicity, oxygen vacancies, redox properties and dispersion of Ni particles, and the understanding of these favorable properties can be beneficial to clarify the catalytic mechanism. Typically, the activation of CH4 and CO2 is considered as a crucial step.53,62 As shown in Fig. 8, the activation of CH4 occurs over the Ni sites, CO2 can be absorbed and activated on the catalyst surface.62 Increasing the basicity of catalysts can increase the rate of activation of CO2, which has a significant influence on the catalytic performance.2 In our study, the addition of rare earth elements can effectively enhance the surface basicity of the catalysts, therefore, the CO2 adsorption capacity of NiMgAl(RE) catalysts can be improved. According to the in situ DRIFTs analysis, the absorbed CO2 can form two kinds of carbonate species on the catalysts surface: bidentate carbonates and monodentate carbonates. Meanwhile, the active intermediate CHx can react with these formed carbonates, especially, the bidentate carbonates are more favorable for CHx conversion. Additionally, the formed carbonates can effectively react with the deposited carbon, and thus, the existence of the carbonates could benefit to the removal of the deposited carbon, leading to the enhanced catalytic stability. | ||
| Fig. 8 Schematic illustration of the possible reaction occurred over the NiMgAlCe catalyst. | ||
Furthermore, the oxygen vacancy and redox property are crucial for improving the catalytic performance. Due to the coexistence of redox pairs, the Ce or Pr modified catalysts showed enhanced redox properties and abundant oxygen vacancies among the NiMgAl(RE) catalysts. The abundant oxygen vacancies can provide supplement active oxygen and more active sites for the activation of CO2 and CH4. Especially, the active oxygen species played an important role in suppressing the carbon deposition. Since the surface active oxygen species could react with the deposited carbon, which contribute to preventing catalyst deactivation during DRM reaction. In addition, the enhanced redox properties can facilitate the electron transport which could promote the rate of CH4/CO2 conversion as well as the elimination of deposited carbon. Besides, the homogeneous dispersion of Ni species showed a positive effect on anti-coking behavior. The confinement effect of LDH precursor can be able to produce highly dispersed Ni particles, and the doped rare earth elements can stabilize the catalysts structure effectively, thus preventing the migration of Ni particles and providing more active Ni sites. The highly dispersed and small Ni nanoparticles could effectively inhibit the carbon nucleation and the subsequent growth, playing an important role in suppressing the coke formation.
4. Conclusions
The comparative catalytic activity and coke resistance were examined in the DRM reaction over NiMgAl(RE) catalysts. We investigated the promoting effect of various rare earth elements (Sc, Y, Ce, and Pr) on the modification of NiMgAl catalysts. While, the catalysts exhibited different catalytic activity and stability depending on the variety of rare earth elements as the promoter. Characterization results revealed that the rare earth elements modified catalysts possess large amount of basic sites to enhance the CO2 adsorption. Compared with unmodified catalysts, the rare earth elements promoted catalysts, especially the Ce or Pr as the promoter, showed the improved catalytic performance in terms of both catalytic stability and coke resistance. The Ce or Pr modified catalysts showed the highest dispersion of Ni particles, which can effectively prevent the migration of Ni species so that the catalysts can achieve relatively stable and the highest conversion of CO2 and CH4. In addition, the Ce or Pr modified catalysts have very quick redox cycling which associate with the creation of oxygen vacancies, contributing to active CO2 and remove carbon deposition. The in situ DRIFTs clarified the details of carbonate species forming during the adsorption of CO2, and the transient studies investigated the dynamic changes of the intermediate species during catalysis processes. The detailed study provides valuable insight for how the doped rare earth elements promote the behavior of the catalysts surface and interface, leading to enhanced catalytic performance.Acknowledgements
The authors acknowledge the support of the National Natural Science Foundation of China (U1462110), the National Basic Research Program of China (973 Program, 2014CB660803). The authors would like to thank Hang Hu for the in situ DRIFTs measurements.References
- G. A. Olah, A. Goeppert, M. Czaun, T. Mathew, R. B. May and G. K. Prakash, J. Am. Chem. Soc., 2015, 137, 8720–8729 CrossRef CAS PubMed.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- D. Baudouin, K. C. Szeto, P. Laurent, A. De Mallmann, B. Fenet, L. Veyre, U. Rodemerck, C. Coperet and C. Thieuleux, J. Am. Chem. Soc., 2012, 134, 20624–20627 CrossRef CAS PubMed.
- D. Baudouin, U. Rodemerck, F. Krumeich, A. d. Mallmann, K. C. Szeto, H. Ménard, L. Veyre, J.-P. Candy, P. B. Webb, C. Thieuleux and C. Copéret, J. Catal., 2013, 297, 27–34 CrossRef CAS.
- S. Das, S. Thakur, A. Bag, M. S. Gupta, P. Mondal and A. Bordoloi, J. Catal., 2015, 330, 46–60 CrossRef CAS.
- T. D. Gould, A. Izar, A. W. Weimer, J. L. Falconer and J. W. Medlin, ACS Catal., 2014, 4, 2714–2717 CrossRef CAS.
- P. Tang, Q. Zhu, Z. Wu and D. Ma, Energy Environ. Sci., 2014, 7, 2580 CAS.
- M. Cargnello, J. J. Delgado Jaen, J. C. Hernandez Garrido, K. Bakhmutsky, T. Montini, J. J. Calvino Gamez, R. J. Gorte and P. Fornasiero, Science, 2012, 337, 713–717 CrossRef CAS PubMed.
- L. Li, L. Zhou, S. Ould-Chikh, D. H. Anjum, M. B. Kanoun, J. Scaranto, M. N. Hedhili, S. Khalid, P. V. Laveille, L. D'Souza, A. Clo and J.-M. Basset, ChemCatChem, 2015, 7, 819–829 CrossRef CAS.
- C.-j. Liu, J. Ye, J. Jiang and Y. Pan, ChemCatChem, 2011, 3, 529–541 CrossRef CAS.
- D. Neagu, T. S. Oh, D. N. Miller, H. Menard, S. M. Bukhari, S. R. Gamble, R. J. Gorte, J. M. Vohs and J. T. Irvine, Nat. Commun., 2015, 6, 8120 CrossRef PubMed.
- M. M. Nair, S. Kaliaguine and F. Kleitz, ACS Catal., 2014, 4, 3837–3846 CrossRef CAS.
- M. H. Amin, K. Mantri, J. Newnham, J. Tardio and S. K. Bhargava, Appl. Catal., B, 2012, 119–120, 217–226 CrossRef CAS.
- Z. Liu, J. Zhou, K. Cao, W. Yang, H. Gao, Y. Wang and H. Li, Appl. Catal., B, 2012, 125, 324–330 CrossRef CAS.
- X. Huang, G. Xue, C. Wang, N. Zhao, N. Sun, W. Wei and Y. Sun, Catal. Sci. Technol., 2016, 6, 449–459 CAS.
- Z. Li, L. Mo, Y. Kathiraser and S. Kawi, ACS Catal., 2014, 4, 1526–1536 CrossRef CAS.
- Y. Ma, X. Wang, X. You, J. Liu, J. Tian, X. Xu, H. Peng, W. Liu, C. Li, W. Zhou, P. Yuan and X. Chen, ChemCatChem, 2014, 6, 3366–3376 CrossRef CAS.
- D. Pakhare, V. Schwartz, V. Abdelsayed, D. Haynes, D. Shekhawat, J. Poston and J. Spivey, J. Catal., 2014, 316, 78–92 CrossRef CAS.
- G. Fan, F. Li, D. G. Evans and X. Duan, Chem. Soc. Rev., 2014, 43, 7040–7066 RSC.
- Y. Zhan, D. Li, K. Nishida, T. Shishido, Y. Oumi, T. Sano and K. Takehira, Appl. Clay Sci., 2009, 45, 147–154 CrossRef CAS.
- X. Du, D. Zhang, L. Shi, R. Gao and J. Zhang, Nanoscale, 2013, 5, 2659–2663 RSC.
- X. Du, D. Zhang, R. Gao, L. Huang, L. Shi and J. Zhang, Chem. Commun., 2013, 49, 6770–6772 RSC.
- A. P. Ferreira, D. Zanchet, J. C. S. Araújo, J. W. C. Liberatori, E. F. Souza-Aguiar, F. B. Noronha and J. M. C. Bueno, J. Catal., 2009, 263, 335–344 CrossRef CAS.
- S. Zhang, S. Muratsugu, N. Ishiguro and M. Tada, ACS Catal., 2013, 3, 1855–1864 CrossRef CAS.
- L. Pino, A. Vita, M. Laganà and V. Recupero, Appl. Catal., B, 2014, 148–149, 91–105 CrossRef CAS.
- O. D. Pavel, B. Cojocaru, E. Angelescu and V. I. Pârvulescu, Appl. Catal., A, 2011, 403, 83–90 CrossRef CAS.
- E. Angelescu, O. D. Pavel, M. Che, R. Bîrjega and G. Constentin, Catal. Commun., 2004, 5, 647–651 CrossRef CAS.
- X. Yu, N. Wang, W. Chu and M. Liu, Chem. Eng. J., 2012, 209, 623–632 CrossRef CAS.
- D. Harshini, D. H. Lee, J. Jeong, Y. Kim, S. W. Nam, H. C. Ham, J. H. Han, T.-H. Lim and C. W. Yoon, Appl. Catal., B, 2014, 148–149, 415–423 CrossRef CAS.
- Z. Xu, N. Wang, W. Chu, J. Deng and S. Luo, Catal. Sci. Technol., 2015, 5, 1588–1597 CAS.
- Z. Wang, P. Fongarland, G. Lu and N. Essayem, J. Catal., 2014, 318, 108–118 CrossRef CAS.
- A. Desmartin-Chomel, B. Hamad, J. Palomeque, N. Essayem, G. Bergeret and F. Figueras, Catal. Today, 2010, 152, 110–114 CrossRef CAS.
- A. R. González, Y. J. O. Asencios, E. M. Assaf and J. M. Assaf, Appl. Surf. Sci., 2013, 280, 876–887 CrossRef.
- R. B. Duarte, M. Nachtegaal, J. M. C. Bueno and J. A. van Bokhoven, J. Catal., 2012, 296, 86–98 CrossRef.
- P. Djinović, I. G. O. Črnivec, B. Erjavec and A. Pintar, ChemCatChem, 2014, 6, 1652–1663 CrossRef.
- M. L. Ang, U. Oemar, E. T. Saw, L. Mo, Y. Kathiraser, B. H. Chia and S. Kawi, ACS Catal., 2014, 4, 3237–3248 CrossRef CAS.
- Y. H. Taufiq-Yap, Sudarno, U. Rashid and Z. Zainal, Appl. Catal., A, 2013, 468, 359–369 CrossRef CAS.
- N. Wang, K. Shen, L. Huang, X. Yu, W. Qian and W. Chu, ACS Catal., 2013, 3, 1638–1651 CrossRef CAS.
- Z. Liu, W. Xu, S. Yao, A. C. Johnson-Peck, F. Zhao, P. Michorczyk, A. Kubacka, E. A. Stach, M. Fernández-García, S. D. Senanayake and J. A. Rodriguez, J. Catal., 2015, 321, 90–99 CrossRef CAS.
- E. W. McFarland and H. Metiu, Chem. Rev., 2013, 113, 4391–4427 CrossRef CAS PubMed.
- S. Li and J. Gong, Chem. Soc. Rev., 2014, 43, 7245–7256 RSC.
- H. Wu, G. Pantaleo, V. La Parola, A. M. Venezia, X. Collard, C. Aprile and L. F. Liotta, Appl. Catal., B, 2014, 156–157, 350–361 CrossRef CAS.
- S. A. Nikolaev and V. V. Smirnov, Catal. Today, 2009, 147, S336–S341 CrossRef CAS.
- K. Sutthiumporn and S. Kawi, Int. J. Hydrogen Energy, 2011, 36, 14435–14446 CrossRef CAS.
- S. S. Maluf and E. M. Assaf, Fuel, 2009, 88, 1547–1553 CrossRef CAS.
- U. Oemar, M. L. Ang, Y. C. Chin, K. Hidajat and S. Kawi, Catal. Sci. Technol., 2015, 5, 3585–3597 CAS.
- S. Radha and A. Navrotsky, J. Phys. Chem. C, 2014, 118, 29836–29844 CAS.
- M. J. Ramírez-Moreno, I. C. Romero-Ibarra, M. A. Hernández-Pérez and H. Pfeiffer, Ind. Eng. Chem. Res., 2014, 53, 8087–8094 CrossRef.
- L. J. I. Coleman, W. Epling, R. R. Hudgins and E. Croiset, Appl. Catal., A, 2009, 363, 52–63 CrossRef CAS.
- T. Odedairo, J. Chen and Z. Zhu, J. Phys. Chem. C, 2013, 117, 21288–21302 CAS.
- M. Yu, K. Zhu, Z. Liu, H. Xiao, W. Deng and X. Zhou, Appl. Catal., B, 2014, 148–149, 177–190 CrossRef CAS.
- M.-S. Fan, A. Z. Abdullah and S. Bhatia, Appl. Catal., B, 2010, 100, 365–377 CrossRef CAS.
- X. Du, D. Zhang, L. Shi, R. Gao and J. Zhang, J. Phys. Chem. C, 2012, 116, 10009–10016 CAS.
- C. Shi and P. Zhang, Appl. Catal., B, 2012, 115–116, 190–200 CrossRef CAS.
- M. García-Diéguez, I. S. Pieta, M. C. Herrera, M. A. Larrubia and L. J. Alemany, J. Catal., 2010, 270, 136–145 CrossRef.
- J. Munera, S. Irusta, L. Cornaglia, E. Lombardo, D. Vargascesar and M. Schmal, J. Catal., 2007, 245, 25–34 CrossRef CAS.
- X. Li, D. Li, H. Tian, L. Zeng, Z.-J. Zhao and J. Gong, Appl. Catal., B, 2017, 202, 683–694 CrossRef CAS.
- X. Zhang, N. Wang, Y. Xu, Y. Yin and S. Shang, Catal. Commun., 2014, 45, 11–15 CrossRef CAS.
- D. Liu, Y. Wang, D. Shi, X. Jia, X. Wang, A. Borgna, R. Lau and Y. Yang, Int. J. Hydrogen Energy, 2012, 37, 10135–10144 CrossRef CAS.
- D. Liu, Y. Wang, D. Shi, X. Jia, X. Wang, A. Borgna, R. Lau and Y. Yang, Int. J. Hydrogen Energy, 2012, 37, 10135–10144 CrossRef CAS.
- S. Wen, M. Liang, J. Zou, S. Wang, X. Zhu, L. Liu and Z.-j. Wang, J. Mater. Chem. A, 2015, 3, 13299–13307 CAS.
- T. Harada, F. Simeon, E. Z. Hamad and T. A. Hatton, Chem. Mater., 2015, 27, 1943–1949 CrossRef CAS.
- J. Guo, Z. Hou, J. Gao and X. Zheng, Chin. J. Catal., 2007, 28, 22–26 CrossRef CAS.
- B. Bachiller-Baeza, C. Mateos-Pedrero, M. A. Soria, A. Guerrero-Ruiz, U. Rodemerck and I. Rodríguez-Ramos, Appl. Catal., B, 2013, 129, 450–459 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Redox property and catalytic stability of NiMgAlCex catalysts containing various amounts of rare earth elements, Ce 3d and Pr 3d XPS spectra of the catalysts, XPS survey scan spectra for NiMgAl(RE) catalysts. In situ DRIFTs of CO2 desorption over the NiMgAlCe catalyst, and the summarization of the peaks in the spectra of CO2 desorption. See DOI: 10.1039/c6ra19139h |
| This journal is © The Royal Society of Chemistry 2016 |