
Green alternative solvents for the copper-catalysed arylation of phenols and amides†
Carlo Sambiagio‡
*a,
Rachel H. Mundayb,
A. John Blackera,
Stephen P. Marsdena and
Patrick C. McGowana
aInstitute for Process Research and Development (iPRD), School of Chemistry, University of Leeds, Woodhouse Lane, LS2 9JT, Leeds, UK
bAstraZeneca Pharmaceutical Development, Hulley Road, SK10 2NA, Macclesfield, UK
First published on 22nd July 2016
Abstract
Investigation of the use of green organic solvents for the Cu-catalysed arylation of phenols and amides is reported. Alkyl acetates proved to be efficient solvents in the catalytic processes, and therefore excellent alternatives to the typical non-green solvents used for Cu-catalysed arylation reactions. Solvents such as isosorbide dimethyl ether (DMI) and diethyl carbonate also appear to be viable possibilities for the arylation of phenols. Finally, a novel copper catalysed acyl transfer process is reported.
Introduction
Metal-catalysed reactions are now essential tools in organic synthesis, and are routinely used even at industrial scale. Industrial applications, however, are subject to more and more strict regulations about waste and toxicity issues. In the field of green chemistry, reaction solvents play an important role, accounting for 50–60% of the material used in a typical process,1,2 and several parameters have been defined to describe their greenness. Large pharmaceutical companies have published tables of solvent data, listing them according to their chemical physical, health and safety, and energy/life-cycle assessment (LCA) properties, leading to banned, dangerous, or recommended classes of solvents for industrial processes.3–7 The number of parameters and their relative importance vary from company to company, affecting the specific preferred solvents and greener alternatives for less green ones. The ACS Green Chemistry Institute-Pharmaceutical Roundtable (ACS GCI-PR) has made available a more general list, developed collaboratively by the members of different pharmaceutical companies,2 and a comparison between individual solvent guides has been published by the Chem21 consortium.8,9Water is always at the top in the lists of green solvents, and much work has been carried out on transition metal-catalysed reactions in this solvent.10–14 However, the physical properties of water do not always make it practically the best solvent for an industrial process,6 and the low solubility of many organic compounds in water represents an obvious issue. Moreover, disposal of large quantities of aqueous waste containing organic and inorganic species requires procedures that are generally far from green.15,16
Copper-catalysed coupling reactions have recently attracted much attention, even at an industrial level, due to the lower toxicity and cost than heavier transition metal catalysts, and considerable research has been undertaken regarding ligands, scope and mechanism.17–20 Green media have been investigated for their use in copper-catalysed arylations since the late 2000s, and now many examples of reactions performed in aqueous solvents can be found in the literature.17,21 Water is frequently used as a co-solvent for these processes in mixtures with DMSO or DMF. When water alone is used, the addition of a phase-transfer catalyst (PTC),22 a polymeric additive,23 or a surfactant24 is often required for effective catalysis. A further disadvantage is the use of hydroxides as bases in these conditions, rather than carbonates or phosphates; these bases are known to be able to react with the aryl halide as nucleophiles, thus leading to the corresponding phenolic side products.17,25 Finally, hydrolysis of particular substituents (e.g. cyano groups) in aqueous conditions has been observed.26,27
As a consequence, the use of organic solvents is still preferred for these reactions, but greener replacements of the traditional non-green solvents need to be investigated. In all of the green solvents lists, water is followed by organic solvents such as alcohols and esters, particularly acetates, both cheap and easily obtained from natural products.3–6 The range of polarities and boiling points also makes these solvents good alternatives to the traditional ones. While alcohols present obvious problems in applications such as C-heteroatom cross couplings, being able to act as competitive nucleophiles (very few examples of copper-catalysed C–N couplings in alcohol media have been reported28,29), acetate solvents may be utilised with less concern. These solvents have been used sporadically for several transition metal-catalysed processes,30–33 including recent Cu-catalysed C–H oxidation processes,34 but no investigation in Cu-catalysed arylations has been reported to date. Other potentially interesting solvents have only been recently added to green solvent tables, being less established.7 Among them, glycerol and its derivatives have been found as good alternatives to other organic solvents in several processes,35–38 and used in agrochemical formulations.39 Derivatives of isosorbide, another naturally-derived compound, have been used as co-solvents or carriers in many pharmaceutical and cosmetic formulations,40,41 thanks to their non-toxicity and favourable chemical properties. Organic carbonates have also attracted much attention, due to their low cost and complete biodegradability, together with their remarkable properties as polar aprotic solvents.42
Herein we report the first investigation on the use of alkyl acetates and other green organic solvents in the copper catalysed arylation of phenols and amides, in combination with picolinamide ligands, which proved effective for the C–O coupling in acetonitrile.43
Results and discussion
Arylation of phenols
The model reaction between 3,5-dimethylphenol and 4-iodoanisole leading to aryl ether 1 was chosen for screening green solvents (Scheme 1). A series of alkyl acetates with different chain lengths were selected, from methyl to pentyl acetate, either linear or branched. Glyceryl triacetate (triacetin), isosorbide dimethyl ether (DMI) and diethyl carbonate (Et2CO3) were also explored as green alternatives.44 A selection of four picolinamide ligands, with different substituents on the phenyl ring, were chosen to study the electronic effects of the ligand in the different solvents. The screening was performed using both Cs2CO3 and K3PO4 as base, which gave the best results in our previous experiments.43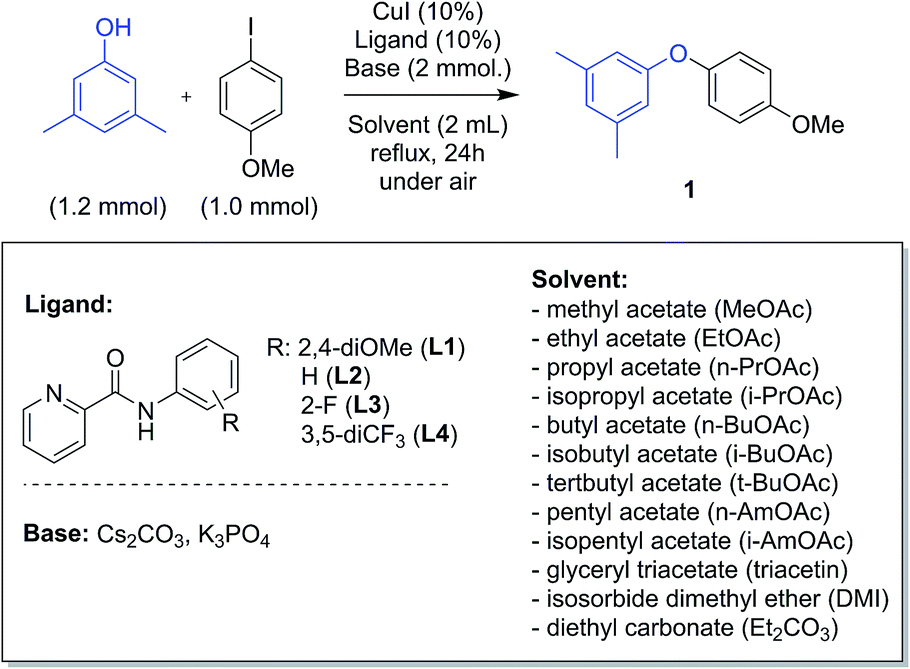 | ||
| Scheme 1 Model reaction for the arylation of phenols in green solvents. | ||
All of the reactions were run under reflux conditions, apart from those in triacetin, due to the exceedingly high boiling point (259 °C); these were run instead at 150 °C. The results of the screening are reported in Fig. 1. The use of Cs2CO3 resulted in generally better yields than K3PO4. In our previous investigation we observed that the catalytic activity increased using ligands bearing electron-withdrawing substituents on the phenyl ring.43 Similar observations can be made for the results in Fig. 1. Ligands L3 or L4, substituted with electron-withdrawing groups are generally more effective than L2 and L1. This is true using either base, but the difference between the ligands is accentuated using K3PO4 (Fig. 1b). In this case, ligand L1 is always considerably less effective than the others, whilst using Cs2CO3 its efficacy is sometimes only a little inferior (Fig. 1a).
 | ||
| Fig. 1 Catalytic results for the synthesis of aryl ether 1: (a) base = Cs2CO3; (b) base = K3PO4 (GC yields using trimethoxybenzene as internal standard). | ||
The previously observed higher activity of the 2-fluoropicolinamide ligand L3 compared to L4 in MeCN, is not generally observed in these solvents, and results with these two ligands are similar. About the different solvents tested, as observed in Fig. 1a, i-PrOAc and i-BuOAc are the best solvents in the acetate series for all the ligands tested, followed by BuOAc, AmOAc and i-AmOAc. The use of PrOAc and t-BuOAc results in poorer yields, and stronger ligand-dependence. The use of shorter chain (methyl and ethyl) acetates resulted in the lowest yields with all ligands.45 Among the other solvents, DMI and Et2CO3 resulted in excellent yields, showing to be interesting alternatives to acetates. The use of triacetin resulted instead in poorer yields, comparable to those observed in MeCN (Fig. 1a). Correlation of the product yields with parameters such as boiling points and dielectric constants for the solvents were checked as a preliminary explanation of these results. While no obvious correlation was observed with the boiling point of the solvents, a general decrease in yields is noted with the increase of the dielectric constant of the reaction solvent (see ESI†).46
A substrate screening was performed to compare the results with those obtained in MeCN (Scheme 2).43 The reactions were performed using ligand L4 and Cs2CO3 in i-PrOAc. This solvent was chosen over the other good ones due to its lower boiling point and consequent milder conditions.47 Isolated yields obtained under these conditions with general electron-donating and electron-withdrawing groups are considerably higher than those obtained in MeCN. Compounds 1–9, with electron-donating to electron-withdrawing substituents on the phenol, were obtained in very good yields, even when electron-rich aryl halides were used. These yields are approx. 20% higher than in MeCN.43 Compound 5 was obtained in 39% yield, much lower than expected for the general trend. This anomalous behaviour was also observed in our previous work.43,48 Reactions with hindered substrates, generally difficult substrates in Cu-catalysed couplings, were also performed. As expected from our previous study, good results were obtained using increasingly bulky substituents (including phenyl) on the phenol moiety in the coupling with the electron-donating 4-iodoanisole (10–13). Steric hindrance on both coupling partners, however, resulted in yields of ca. 20% (14–17). The use of 2,6-dimethylphenol, hindered at both sides, considerably reduces the reactivity, and coupling products with 4-iodoanisole and 2-iodotoluene were obtained in 37% and 22% yield respectively (18–19). The coupling of 3,5-dimethylphenol with 2-iodotoluene furnished 20 in 45% yield, but the reaction with 4-iodoaniline or 2-iodoaniline afforded 79% and 71% yield respectively (21–22). No competing arylation of the amino group was observed in these cases. Compound 23 was obtained in 17% yield, while arylation of 8-hydroxyquinaldine (24) occurred in 21% yield (in MeCN: 4% and 37% respectively43). Aryl bromides were also tested in the reaction: reaction with 4-bromoanisole resulted in max 40% yield, considerably lower than the corresponding iodide (compounds 1, 4, 7, 10, 21).
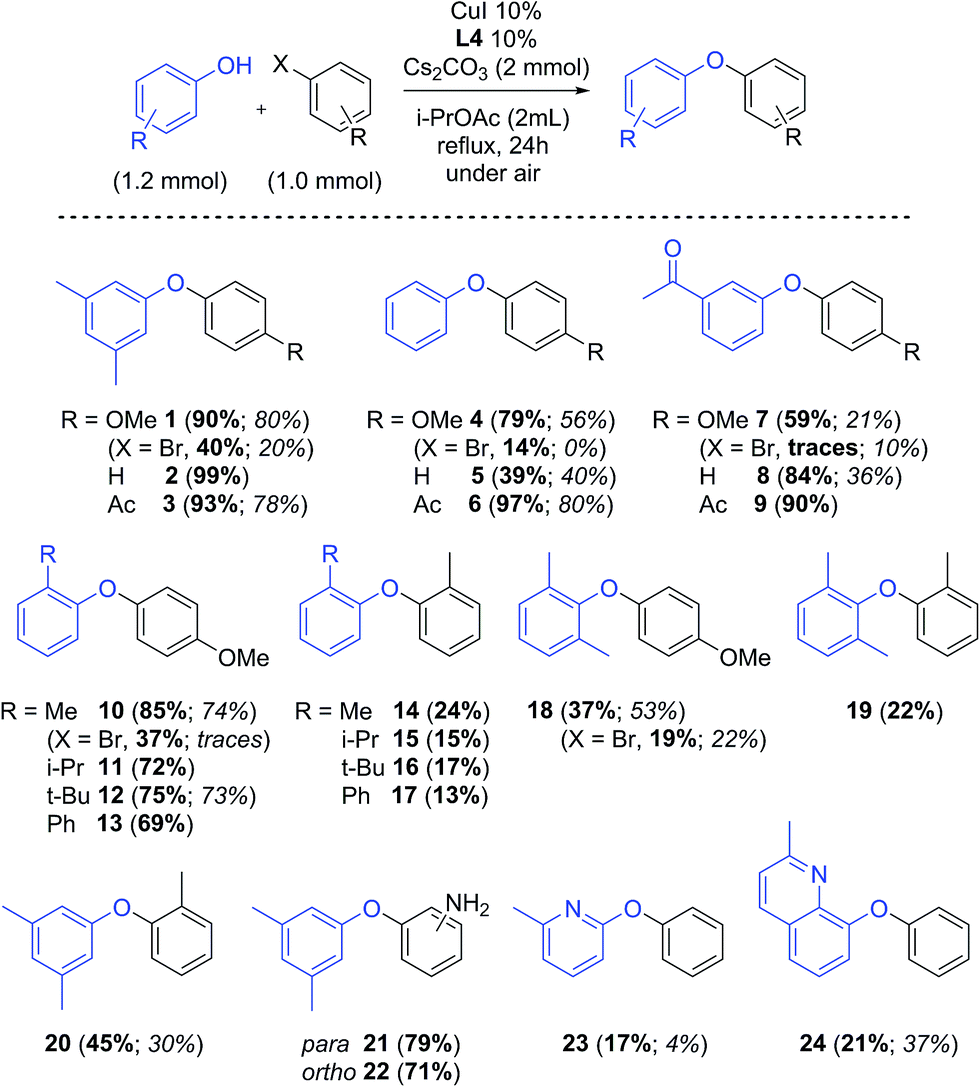 | ||
| Scheme 2 Substrate scope: isolated yields in i-PrOAc (bold) and MeCN (italics, from ref. 43); X = I, if not otherwise specified. | ||
Arylation of amides
The promising results obtained in the arylation of phenols prompted us to investigate whether the arylation of amides was also possible in these solvents. Although a few specific applications of picolinamide ligands in the N-arylation of uracyl can be found in the literature, no detailed study has been reported on the use of these ligands for amide arylation.49,50 Benzamide and 4-iodoanisole were chosen as model substrates for the arylation of amides (Scheme 3). The results obtained for these couplings with L1–L4 in the solvents discussed above are shown in Fig. 2. In this case, the use of Cs2CO3 as base resulted in generally poor yields, and not defined ligand effect could be clearly distinguished. The use of K3PO4 instead resulted in cleaner reactions, higher yields, and more sensible trends, with electron-withdrawing substituted picolinamide ligands more effective in the catalysis (Fig. 2b). Norm- and iso-butyl and pentyl acetates proved effective solvents for the synthesis of benzamide 25, whereas shorter-chain acetates and t-BuOAc acetates were considerably less effective. DMI and diethylcarbonate also resulted in lower yields.51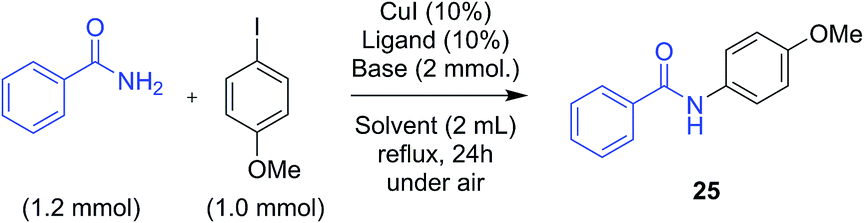 | ||
| Scheme 3 Model reaction for the arylation of amides in green solvents. | ||
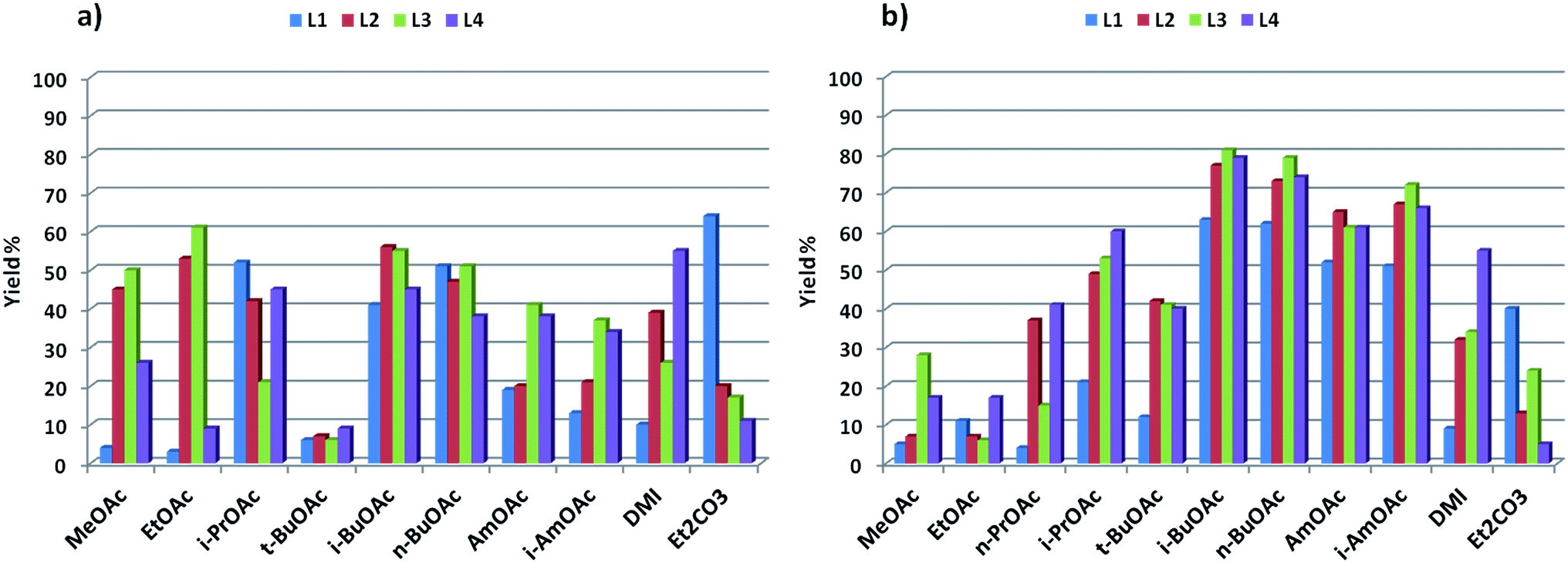 | ||
| Fig. 2 Catalytic results for the synthesis of amide 25: (a) base = Cs2CO3; (b) base = K3PO4 (1H-NMR yields using trimethoxybenzene as internal standard). | ||
i-BuOAc and 2-fluoropicolinamide ligand L3, which furnished the best results in the screening, were chosen to investigate the substrate scope of this methodology (Scheme 4). A range of amides, including aromatic, aliphatic and cyclic amides were explored, resulting in good to excellent yields of the arylated products. The reaction of benzamide, 4-methoxy benzamide and 3-(trifluoromethyl)benzamide with electron-donating to electron-withdrawing aryl iodides occurred with good yields (25–32, 56% to 91% yields). Arylation of cyclohexanecarboxamide gave products 33–35 with yields between 71% and 78%, while cyclic amides such as 2-pyrrolidinone and 2-hydroxypyridine gave excellent results, with yields of 87–98% (36–40). Arylation of nicotinamide with iodobenzene afforded 61% yield of amide 41, while compound 42 was obtained in modest yield (31%). Finally the use of sterically hindered 2-iodotoluene as coupling partner resulted in a considerable decrease in yield, giving products 43–46 in 36–63% yields. Interestingly, 81% yield from the coupling with 2-pyrrolidinone was obtained (47), suggesting a lower steric hindrance sensitivity for this substrate. The yields obtained for the compounds compare well with previously reported results with traditional solvents.52–54
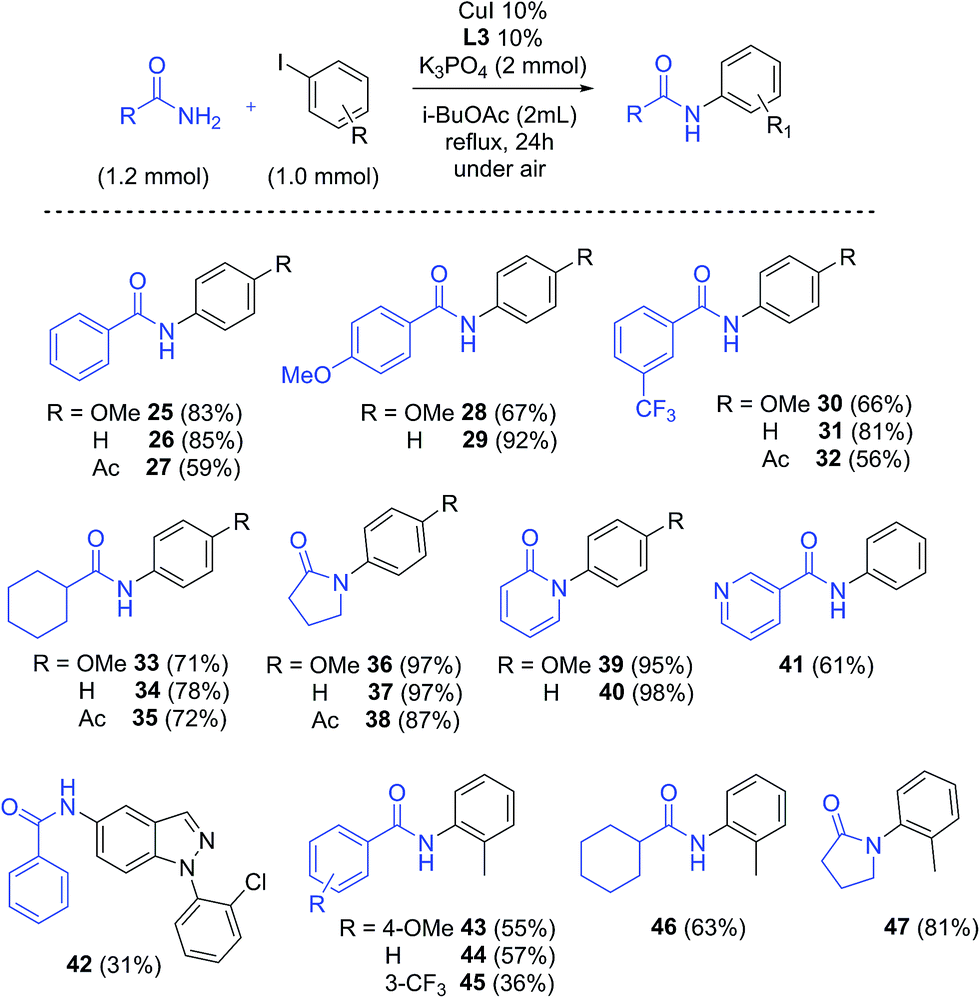 | ||
| Scheme 4 Substrate scope: isolated yields in i-BuOAc. | ||
Benzylation also proved effective under these conditions: from the reaction of benzamide with 2-iodobenzyl bromide, a doubly substituted product was obtained in 30% yield (48, Scheme 5), while the coupling with benzyl bromide and benzyl chloride occurred with 52% and 33% yields respectively (49). It is worth noting that product 49 was not observed in reactions without CuI/ligand, either with benzyl bromide or benzyl chloride.
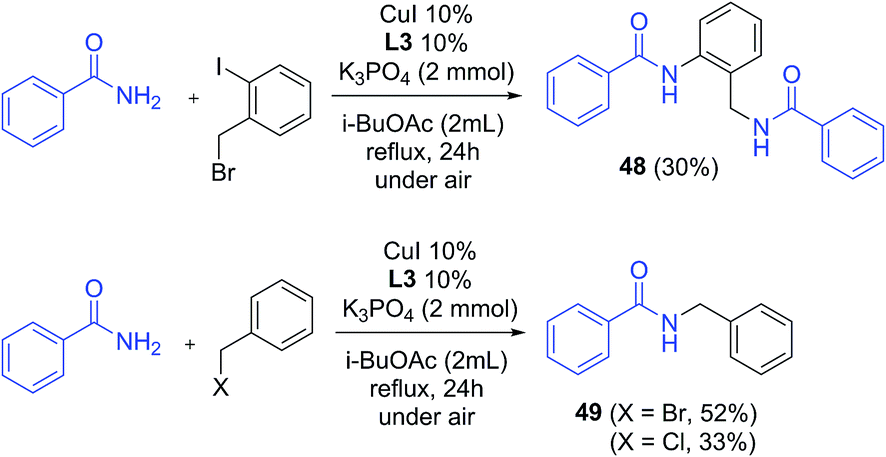 | ||
| Scheme 5 Benzylation of benzamide (isolated yields, yield of 48 based on the amide). | ||
Acyl transfer as a side process
Contrarily to the arylation of phenols, where no side reactions were observed, the arylation of aromatic amides was accompanied by the formation, generally in small amounts, of a side product derived by the reaction of the starting amide with the solvent to form the corresponding isobutyl ester (Scheme 6, compounds 50–53, this process did not occur with cyclohexanecarboxamide). The yields of 50–53 isolated during this study are reported in Table 1.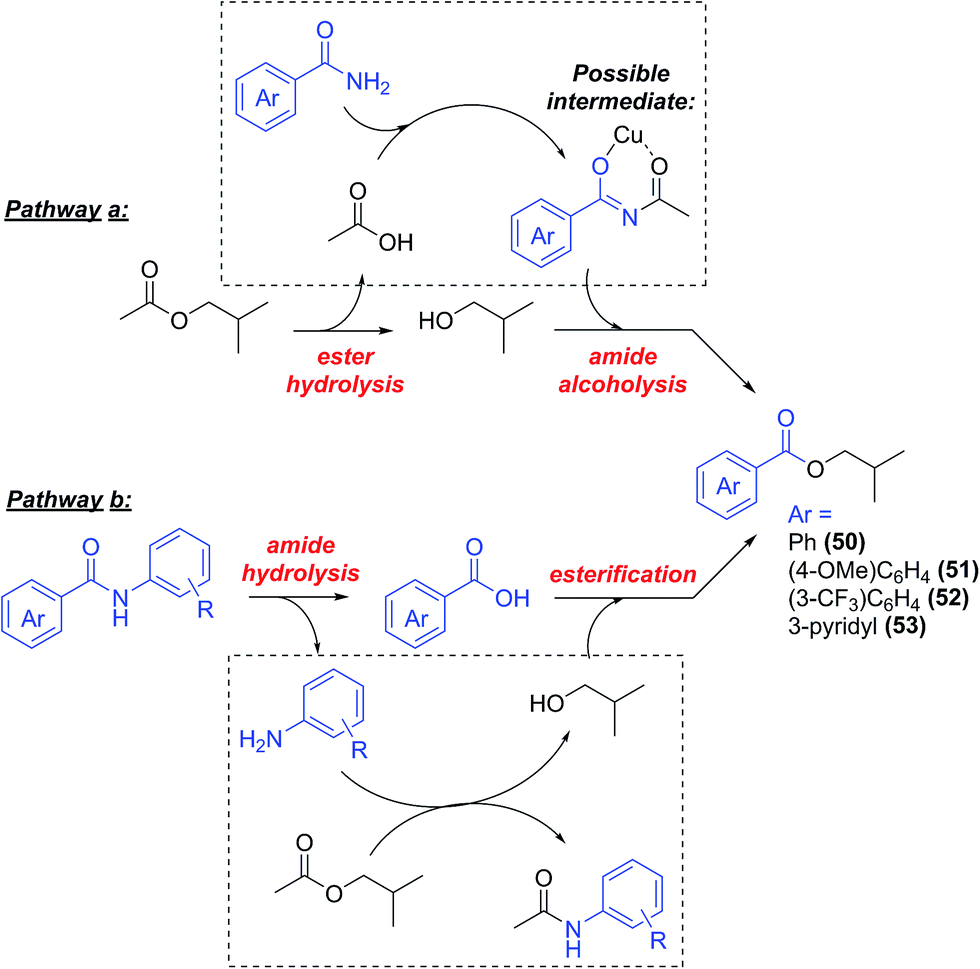 | ||
| Scheme 6 Possible pathways to the formation of side products 50–53. | ||
Very recently, an analogous acyl–acyl exchange between aromatic amides and esters was reported by Bian and co-workers. The process proved to be base-catalysed (K2CO3 was used as base), and did not require any transition metal catalyst.55 However, only aromatic esters (apart from one case) were reported to react in this way, moreover, nicotinamide did not prove a successful substrate in the author's conditions, while a small amount of compound 53 was observed in our case. In a control reaction between benzamide and i-BuOAc in the experimental conditions employed in this study (2 eq. K3PO4, 24 h at reflux temp.) in the absence of CuI/ligand, no isobutyl benzoate 50 was observed (Scheme 7a), suggesting that a metal catalysed process is operative in our conditions.
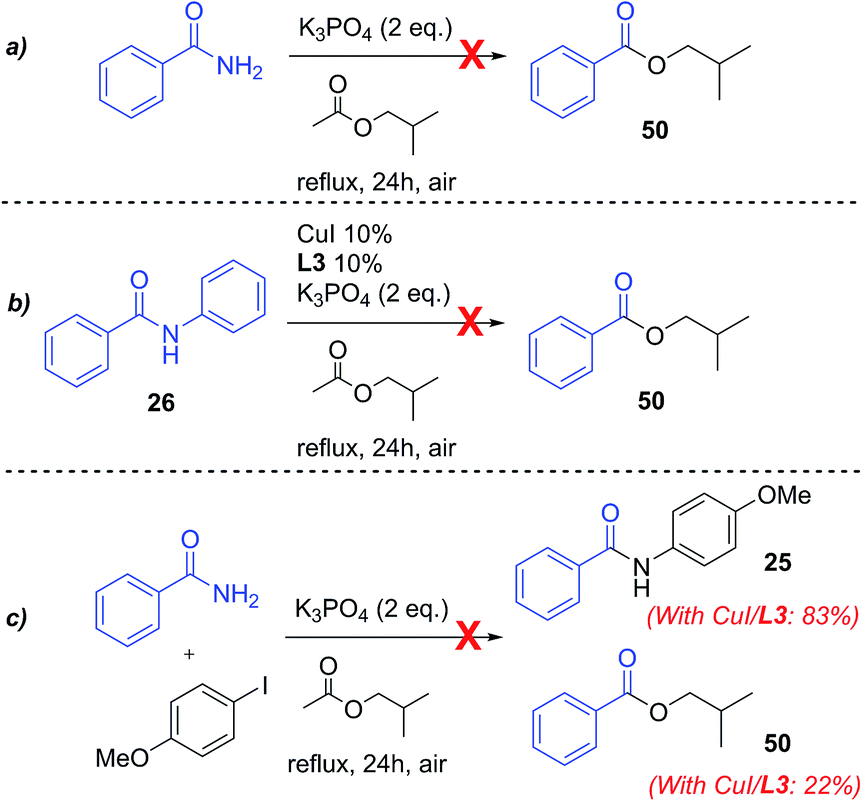 | ||
| Scheme 7 Formation of isobutylbenzoates: control reactions. | ||
These products can in principle be formed through a tandem process involving hydrolysis of i-BuOAc and successive amide alcoholysis (Scheme 6, pathway a). Cu-catalysed hydrolysis of carboxylic esters56–59 is known, and no free isobutyl alcohol was observed in the commercial solvent used (see ESI†), so its hydrolysis would also, likely, be due to the presence of the copper catalyst. The alcoholysis of amides, in contrast, is a difficult process, and only few metal-free procedures exist in the literature.60,61 Berreau and Rivas reported the use of group 12 metal complexes with polydentate ligands for this reaction, mimicking the active site of peptidase enzymes,62,63 showing that coordination of the metal to the carbonyl oxygen facilitates the nucleophilic attack of the alkoxide, leading to the formation of the ester. The same activation may be at the basis of the amide alcoholysis catalysed by simple transition metal salts64–68 or lanthanides salts and oxides.64,69,70 Some examples of Cu-mediated alcoholysis of coordinated amides (i.e. in pre-formed Cu complexes), have been reported by Brown and co-workers.71–73 In our case, the hydrolysis of i-BuOAc may be followed by N-acylation of the benzamide, leading to an imide intermediate, thus facilitating the nucleophilic attack by the isobutoxide ion, and the formation of the benzoic esters 50–53. Further activation of such intermediate might be suggested to occur through coordination to the copper salt (Scheme 6). A similar chelated intermediate has been recently suggested to form during the hydrodeoxygenation of lignin, upon addition of Zn(II) salts in solution. In this case, the process results in the cleavage of a C–O bond.74 Another possible pathway would be the hydrolysis of the final N-arylamide to aniline and benzoic acid (as Cu-catalysed amide hydrolysis is known75–77), followed by reaction with i-BuOAc to give acetanilide and the corresponding benzoic esters (Scheme 6, pathway b). However, substituted acetanilides were not observed in the reaction mixtures, and no hydrolysis of the structurally analogous picolinamide ligands used in this work was observed. A control reaction between benzanilide 26 and i-BuOAc in the catalytic conditions, only showed the presence of the starting material in the H-NMR spectrum (Scheme 7b, and ESI†), thus disproving this reaction pathway.78
Finally, the coupling between benzamide and 4-iodoanisole, performed in the absence of copper and ligand, did not result in any arylation or acyl transfer, and only starting materials were recovered (Scheme 7c and ESI†), proving both processes to be metal-catalysed. Beside these experiments, a control reaction in the absence of 4-iodoanisole, also did not give the isobutylbenzoate product 50, as well as the reaction between benzamide and isobutyl alcohol in the catalytic conditions. While in this last case solubility and mixing problems were observed, resulting in non-reliable data, the negative results obtained for the reaction in the absence of iodoanisole is not readily explainable at the moment. Although further mechanistic investigation would be needed to ascertain the mechanism of this transformation, pathway a depicted in Scheme 6 represents a plausible process, and consistent with previous literature data, being both processes known to occur in Cu-catalysed/mediated conditions. To our knowledge no Cu-catalysed amide alcoholysis has been reported before.
Conclusions
In conclusion, we reported the first systematic study on the use on green organic solvents for the Cu-catalysed arylation of phenols and amides. Our results show that alkyl acetates are viable alternatives to the standard organic solvents used in copper-catalysed cross couplings. In particular, the yields for the synthesis of aryl ethers in i-PrOAc were higher than those obtained in acetonitrile, while the general group and steric hindrance tolerance remained unchanged. A range of different amides (aromatic, aliphatic and cyclic amides) can also be effectively arylated in i-BuOAc using K3PO4 as a base. The use of these solvents, considerably greener that the traditional solvents used for copper-catalysed C–O and C–N bond formation, might be of interest for industry-based and process chemistry research, and we hope this study will prompt more investigation on the use of such solvents in other metal-catalysed reactions. Finally, a novel Cu-catalysed acyl transfer process between aromatic amides and alkyl acetates has been discovered.Experimental section
General
NMR spectra were acquired using a Bruker Avance 500 MHz spectrometer. Chemical shifts (δ) are expressed in ppm and referred to the solvent signal (CDCl3: 7.27 ppm (H) and 77.0 ppm (C)). Mass spectra were recorded by the author at the University of Leeds Mass Spectrometry Service on a Bruker Daltonics MicroTOF instrument. Purification by column chromatography was carried out using Merck Geduran Si 60 Silicagel.General procedure for the synthesis of picolinamide ligands
In a round-bottom flask equipped with a condenser, 1 eq. of picolinic acid was stirred with pyridine (min. 1 eq.) at room temperature, then 1 eq. of aniline and 1 eq. of triphenyl phosphite were added to the solution. The mixture was heated to reflux and stirred for 16 h. At the end of the reaction the solution was left to cool to room temperature and an excess of distilled water was added, stirring the mixture for an additional hour. The crude solid product, precipitated after the addition of water, was purified by recrystallisation from methanol.General procedure for the synthesis of aryl ethers
All catalytic reactions were performed under air in 25 mL glass tubes in a Radleys standard carousel. An oven dried tube was charged with the phenol (1.2 mmol), the aryl iodide (1.0 mmol), caesium carbonate (652 mg, 2 mmol), CuI (19 mg, 0.1 mmol) and ligand (0.1 mmol). Isopropyl acetate (2 mL) was then added, the tube was placed in the carousel (preheated at 110 °C) and the reaction was stirred for 24 h (1000 rpm). At the end of the reaction the tube was cooled to room temperature, then the crude was diluted with DCM, filtered through celite and washed with DCM (≈50 mL). Isolated compounds were purified by column chromatography. Chemicals used for the initial screening: Sigma Aldrich CuI (98%); Alfa Aesar Cs2CO3 (99+% metal basis), Alfa Aesar K3PO4 (97%, anhydrous, granular); Alfa Aesar 4-iodoanisole (98+%); Acros Organics 3,5-dimethylphenol (99+%). Solvents: Sigma Aldrich MeOAc (for HPLC, ≥99.8%); VWR Prolabo EtOAc (GPR Rectapur, min. 99.0%); Sigma Aldrich n-PrOAc (99%); Alfa Aesar i-PrOAc (99+%); Sigma Aldrich n-BuOAc (anhydrous, ≥99%); Sigma Aldrich t-BuOAc (≥99%); Alfa Aesar i-BuOAc (98%); Sigma Aldrich n-AmOAc (99%); Sigma Aldrich i-AmOAc (reagent grade, 98%); Merck isosorbide dimethyl ether (for synthesis, ≥98%); Sigma Aldrich glyceryl triacetate (>99%); Sigma Aldrich diethyl carbonate (>99%).General procedure for the synthesis of amides
All catalytic reactions were performed under air in 25 mL glass tubes in a Radleys standard carousel. An oven dried tube was charged with the amide (1.2 mmol), the aryl iodide (1.0 mmol), potassium phosphate (424 mg, 2 mmol), CuI (19 mg, 0.1 mmol) and ligand (0.1 mmol). Isobutyl acetate (2 mL) was then added, the tube was placed in the carousel (preheated at 150 °C) and the reaction was stirred for 24 h (1000 rpm). At the end of the reaction the tube was cooled to room temperature, then the crude was diluted with DCM, filtered through celite and washed with DCM (≈50 mL). Isolated compounds were purified by column chromatography. Chemicals used for the initial screening: Sigma Aldrich CuI (98%); Alfa Aesar Cs2CO3 (99+% metal basis), Alfa Aesar K3PO4 (97%, anhydrous, granular); Alfa Aesar 4-iodoanisole (98+%); Sigma Aldrich benzamide (99%). Solvents: Sigma Aldrich MeOAc (for HPLC, ≥99.8%); VWR Prolabo EtOAc (GPR Rectapur, min 99.0%); Sigma Aldrich n-PrOAc (99%); Alfa Aesar i-PrOAc (99+%); Sigma Aldrich n-BuOAc (anhydrous, ≥99%); Sigma Aldrich t-BuOAc (≥99%); Alfa Aesar i-BuOAc (98%); Sigma Aldrich n-AmOAc (99%); Sigma Aldrich i-AmOAc (reagent grade, 98%); Merck isosorbide dimethyl ether (for synthesis, ≥98%); Sigma Aldrich diethyl carbonate (>99%).Acknowledgements
We are thankful to AstraZeneca UK and the University of Leeds for an Industrial CASE studentship (C. S.). We are also grateful to Drs David Hose and Louis Diorazio (AZ) for useful comments and discussion.Notes and references
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- ACS GCI-PR solvent selection guide 2011, http://www.acs.org/content/dam/acsorg/greenchemistry/industriainnovation/roundtable/acs-gci-pr-solvent-selection-guide.pdf, accessed 16 Jan., 2016.
- AZ solvent selection guide has not been published, but has been reported in the following presentation, http://www.acs.org/content/dam/acsorg/greenchemistry/industriainnovation/roundtable/solvent-selection-guide.pdf, accessed 16 Jan., 2016.
- R. K. Henderson, C. Jimenez-Gonzalez, D. J. C. Constable, S. R. Alston, G. G. A. Inglis, G. Fisher, J. Sherwood, S. P. Binks and A. D. Curzons, Green Chem., 2011, 13, 854–862 RSC.
- K. Alfonsi, J. Colberg, P. J. Dunn, T. Fevig, S. Jennings, T. A. Johnson, H. P. Kleine, C. Knight, M. A. Nagy, D. A. Perry and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC.
- D. Prat, O. Pardigon, H.-W. Flemming, S. Letestu, V. Ducandas, P. Isnard, E. Guntrum, T. Senac, S. Ruisseau, P. Cruciani and P. Hosek, Org. Process Res. Dev., 2013, 17, 1517–1525 CrossRef CAS.
- C. M. Alder, J. D. Hayler, R. K. Henderson, A. M. Redman, L. Shukla, L. E. Shuster and H. F. Sneddon, Green Chem., 2016, 18, 3879–3890 RSC.
- D. Prat, J. Hayler and A. Wells, Green Chem., 2014, 16, 4546–4551 RSC.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- H. D. Velazquez and F. Verpoort, Chem. Soc. Rev., 2012, 41, 7032–7060 RSC.
- B. Li and P. H. Dixneuf, Chem. Soc. Rev., 2013, 42, 5744–5767 RSC.
- B. H. Lipshutz and S. Ghorai, Aldrichimica Acta, 2012, 45, 3–16 CAS.
- A. Postigo and N. S. Nudelman, Coord. Chem. Rev., 2011, 255, 2991–3030 CrossRef CAS.
- R. N. Butler and A. G. Coyne, Chem. Rev., 2010, 110, 6302–6337 CrossRef CAS PubMed.
- D. G. Blackmond, A. Armstrong, V. Coombe and A. Wells, Angew. Chem., Int. Ed., 2007, 46, 3798–3800 CrossRef CAS PubMed.
- D. J. C. Constable, C. Jimenez-Gonzalez and R. K. Henderson, Org. Process Res. Dev., 2007, 11, 133–137 CrossRef CAS.
- C. Sambiagio, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem. Soc. Rev., 2014, 43, 3525–3550 RSC.
- D. S. Surry and S. L. Buchwald, Chem. Sci., 2010, 1, 13–31 RSC.
- F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 6954–6971 CrossRef CAS PubMed.
- D. Ma and Q. Cai, Acc. Chem. Res., 2008, 41, 1450–1460 CrossRef CAS PubMed.
- M. Carril, R. SanMartin and E. Dominguez, Chem. Soc. Rev., 2008, 37, 639–647 RSC.
- D. Wang, F. Zhang, D. Kuang, J. Yu and J. Li, Green Chem., 2012, 14, 1268–1271 RSC.
- J. Engel-Andreasen, B. Shimpukade and T. Ulven, Green Chem., 2013, 15, 336–340 RSC.
- S. Liu and J. Zhou, New J. Chem., 2013, 37, 2537–2540 RSC.
- A. Tlili, N. Xia, F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 8725–8728 CrossRef CAS PubMed.
- S. Maurer, W. Liu, X. Zhang, Y. Jiang and D. Ma, Synlett, 2010, 2010, 976–978 CrossRef.
- Y. Xiao, Y. Xu, H.-S. Cheon and J. Chae, J. Org. Chem., 2013, 78, 5804–5809 CrossRef CAS PubMed.
- F. Y. Kwong, A. Klapars and S. L. Buchwald, Org. Lett., 2002, 4, 581–584 CrossRef CAS PubMed.
- A. Sharifi, N. Ghonouei and M. S. Abaee, Synth. Commun., 2016, 46, 249–256 CrossRef CAS.
- C. J. Pink, H.-t. Wong, F. C. Ferreira and A. G. Livingston, Org. Process Res. Dev., 2008, 12, 589–595 CrossRef CAS.
- Akanksha and D. Maiti, Green Chem., 2012, 14, 2314–2320 RSC.
- A. Mouret, L. Leclercq, A. Muhlbauer and V. Nardello-Rataj, Green Chem., 2014, 16, 269–278 RSC.
- R. Savela, J. Warna, D. Y. Murzin and R. Leino, Catal. Sci. Technol., 2015, 5, 2406–2417 CAS.
- H. Sterckx, J. De Houwer, C. Mensch, W. Herrebout, K. A. Tehrani and B. U. W. Maes, Beilstein J. Org. Chem., 2016, 12, 144–153 CrossRef CAS PubMed.
- P. Cintas, S. Tagliapietra, E. Calcio Gaudino, G. Palmisano and G. Cravotto, Green Chem., 2014, 16, 1056–1065 RSC.
- C. Vidal and J. Garcia-Alvarez, Green Chem., 2014, 16, 3515–3521 RSC.
- S. Thurow, F. Penteado, G. Perin, R. G. Jacob, D. Alves and E. J. Lenardao, Green Chem., 2014, 16, 3854–3859 RSC.
- A. Wolfson, A. Snezhko, T. Meyouhas and D. Tavor, Green Chem. Lett. Rev., 2011, 5, 7–12 CrossRef.
- G. Schnabel, WO 2013153030 A1, 2013.
- M.-K. Tran, A. Swed and F. Boury, Eur. J. Pharm. Biopharm., 2012, 82, 498–507 CrossRef CAS PubMed , and references therein.
- Y. Zhu, M. Durand, V. Molinier and J.-M. Aubry, Green Chem., 2008, 10, 532–540 RSC.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554–4581 CrossRef PubMed , and references therein.
- C. Sambiagio, R. H. Munday, S. P. Marsden, A. J. Blacker and P. C. McGowan, Chem.–Eur. J., 2014, 20, 17606–17615 CrossRef CAS PubMed.
- Ethyl lactate as solvent was also tested, but it resulted in all cases in a sticky gel-like product, insoluble in different solvents, leading to difficult workup and non-reliable results.
- The much higher yield obtained with 2-fluoropicolinamide L3 in EtOAc with Cs2CO3 has been reproduced (within 10%).
- Dielectric constant values for i-PrOAc, t-BuOAc and DMI could not be found in the literature or the web, making the correlation impossible for these solvents at this stage.
- Note, however, that i-BuOAc may be preferred from a process point of view, due to its higher boiling point.
- In our previous study we observed that, using MeCN, reactions involving unsubstituted phenol resulted, at the end of the reaction, in a quantity of red/lilac solid that was filtered off. Although we were not able to identify it, we suggested this could be related to some metal phenoxide species. Such colour was however not observed for the reactions in i-PrOAc.
- A. C. Krueger, J. T. Randolph, D. A. DeGoey, P. L. Donner, C. A. Flentge, D. K. Hutchinson, D. Liu, C. E. Motter, T. W. Rockway, R. Wagner, D. W. A. Beno, G. Koev, H. B. Lim, J. M. Beyer, R. Mondal, Y. Liu, W. M. Kati, K. L. Longenecker, A. Molla, K. D. Stewart and C. J. Maring, Bioorg. Med. Chem. Lett., 2013, 23, 3487–3490 CrossRef CAS PubMed.
- F. X. Talamas, S. C. Abbot, S. Anand, K. A. Brameld, D. S. Carter, J. Chen, D. Davis, J. de Vicente, A. D. Fung, L. Gong, S. F. Harris, P. Inbar, S. S. Labadie, E. K. Lee, R. Lemoine, S. Le Pogam, V. Leveque, J. Li, J. McIntosh, I. Nájera, J. Park, A. Railkar, S. Rajyaguru, M. Sangi, R. C. Schoenfeld, L. R. Staben, Y. Tan, J. P. Taygerly, A. G. Villaseñor and P. E. Weller, J. Med. Chem., 2014, 57, 1914–1931 CrossRef CAS PubMed.
- n-PrOAc resulted in insoluble sticky solid at the bottom of the reaction tubes, when using Cs2CO3 as base. This made the results unreliable, and are not reported in Fig. 2a. Similar results were obtained for the reactions in triacetin using both bases.
- H.-J. Cristau, P. P. Cellier, J.-F. Spindler and M. Taillefer, Chem.–Eur. J., 2004, 10, 5607–5622 CrossRef CAS PubMed.
- Z.-J. Quan, H.-D. Xia, Z. Zhang, Y.-X. Da and X.-C. Wang, Tetrahedron, 2013, 69, 8368–8374 CrossRef CAS.
- W. Deng, Y.-F. Wang, Y. Zou, L. Liu and Q.-X. Guo, Tetrahedron Lett., 2004, 45, 2311–2315 CrossRef CAS.
- Y. Bian and X. Qu, Org. Biomol. Chem., 2016, 14, 3869–3872 CAS.
- R. Breslow and B. Zhang, J. Am. Chem. Soc., 1992, 114, 5882–5883 CrossRef CAS.
- B. E. Leach and R. J. Angelici, Inorg. Chem., 1969, 8, 907–913 CrossRef CAS.
- J. G. J. Weijnen, A. Koudijs and J. F. J. Engbersen, J. Chem. Soc., Perkin Trans. 2, 1991, 1121–1126 RSC.
- R. Nakon, P. R. Rechani and R. J. Angelici, J. Am. Chem. Soc., 1974, 96, 2117–2120 CrossRef CAS PubMed.
- D. F. Taber and M. Rahimizadeh, J. Org. Chem., 1992, 57, 4037–4038 CrossRef CAS.
- A. J. Kiessling and C. K. McClure, Synth. Commun., 1997, 27, 923–937 CrossRef CAS.
- E. Szajna, M. M. Makowska-Grzyska, C. C. Wasden, A. M. Arif and L. M. Berreau, Inorg. Chem., 2005, 44, 7595–7605 CrossRef CAS PubMed.
- J. C. M. Rivas, E. Salvagni and S. Parsons, Dalton Trans., 2004, 4185–4192 RSC.
- A. A. Neverov, P. J. Montoya-Pelaez and R. S. Brown, J. Am. Chem. Soc., 2001, 123, 210–217 CrossRef CAS PubMed.
- C. T. Liu, C. I. Maxwell, S. G. Pipe, A. A. Neverov, N. J. Mosey and R. S. Brown, J. Am. Chem. Soc., 2011, 133, 20068–20071 CrossRef CAS PubMed.
- B. N. Atkinson and J. M. J. Williams, Tetrahedron Lett., 2014, 55, 6935–6938 CrossRef CAS.
- Y. Kita, Y. Nishii, A. Onoue and K. Mashima, Adv. Synth. Catal., 2013, 355, 3391–3395 CrossRef CAS.
- Y. Kita, Y. Nishii, T. Higuchi and K. Mashima, Angew. Chem., Int. Ed., 2012, 51, 5723–5726 CrossRef CAS PubMed.
- M. de Léséleuc and S. K. Collins, ACS Catal., 2015, 5, 1462–1467 CrossRef.
- S. M. A. H. Siddiki, A. S. Touchy, M. Tamura and K.-i. Shimizu, RSC Adv., 2014, 4, 35803–35807 RSC.
- I. F. Barrera, C. I. Maxwell, A. A. Neverov and R. S. Brown, J. Org. Chem., 2012, 77, 4156–4160 CrossRef CAS PubMed.
- M. A. R. Raycroft, L. Cimpean, A. A. Neverov and R. S. Brown, Inorg. Chem., 2014, 53, 2211–2221 CrossRef CAS PubMed.
- M. A. R. Raycroft, C. I. Maxwell, R. A. A. Oldham, A. S. Andrea, A. A. Neverov and R. S. Brown, Inorg. Chem., 2012, 51, 10325–10333 CrossRef CAS PubMed.
- I. Klein, C. Marcum, H. Kenttamaa and M. M. Abu-Omar, Green Chem., 2016, 18, 2399–2405 RSC.
- K. V. Reddy, A. R. Jacobson, J. I. Kung and L. M. Sayre, Inorg. Chem., 1991, 30, 3520–3525 CrossRef CAS.
- L. M. Sayre, K. V. Reddy, A. R. Jacobson and W. Tang, Inorg. Chem., 1992, 31, 935–937 CrossRef CAS.
- N. N. Murthy, M. Mahroof-Tahir and K. D. Karlin, J. Am. Chem. Soc., 1993, 115, 10404–10405 CrossRef CAS.
- This pathway was suggested by a referee of the original manuscript. Although our results suggest that the acyl transfer does not occur with this mechanism, this possibility is worth keeping in mind for further mechanistic studies.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional figures, characterisation data and copies of NMR spectra for all isolated products. See DOI: 10.1039/c6ra02265k |
| ‡ Current address: Department of Chemistry, University of Antwerp, Groenenborgerlaan 171, B-2020, Antwerp, Belgium, E-mail: E-mail: Carlo.Sambiagio@uantwerpen.be |
| This journal is © The Royal Society of Chemistry 2016 |