
Regioregularity-control of conjugated polymers: from synthesis and properties, to photovoltaic device applications

Abstract
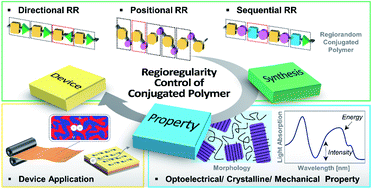
In the last few decades, extensive academic and industrial efforts have been devoted to developing high-performance conjugated polymers (CPs) for organic electronics. Specifically, the relationships between the molecular structures of CPs and their properties and device characteristics have been a subject of intense studies. In this review, we highlight recent advances in the molecular design of CPs, particularly on tuning their regioregularity (RR). The RR of repeating units (i.e., directional, positional, and sequential regularities) within the CP backbone determines the intrinsic properties of CPs and the performances of the resulting devices. Despite the significant impact of RR on the overall properties of the polymers and the device performances, the importance of RR in the design of CPs has yet to be emphasized. Furthermore, RR control is critical in state-of-the-art CPs with asymmetric molecular structures. This review presents a library of examples that report the impact of RR control in CPs on the properties and performances in various organic electronic devices, including simple homopolymers with asymmetric alkyl side chains and more recently developed copolymers with various donor–acceptor (D–A) combinations. This review summarizes important guidelines and provides insights for the molecular design of RR-controlled CPs and their applications in efficient organic electronics.
- This article is part of the themed collection: 2023 Journal of Materials Chemistry A Lunar New Year collection
 Please wait while we load your content...
Please wait while we load your content...

