
Interpenetrated triple network polymers: synergies of three different dynamic bonds†

Abstract
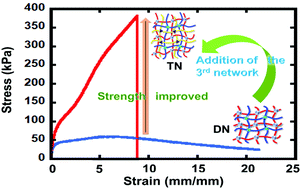
An ongoing challenge in soft materials is to develop networks with high mechanical robustness while showing complete self-healing and stress relaxation. In this study we develop triple network (TN) materials with three different polymers with distinct dynamic linkers (Diels–Alder, boronic acid-ester and hydrogen bonding). TN materials exhibit significant improvement of strength, stability and excellent self-healing properties simultaneously compared to their analogous double networks (DNs). All the TNs (TN-FMA 5%, 7% and 9%) show higher tensile strength over all DNs. In addition, TN-FMA (9%) demonstrates an excellent fracture energy over 20 000 J m−2, 750% elongation and fast stress relaxation. This highlights how dynamic bonding multiplicity and network structure can play a major role in improving the quality of dynamic materials.
- This article is part of the themed collections: Polymer Chemistry Lectureship Winners and Polymer Networks
 Please wait while we load your content...
Please wait while we load your content...

