
Extraordinary magnetic field effects on the LC phases of homochiral and racemic 4-cyanoresorcinol-based diamagnetic bent-core mesogens†

Abstract
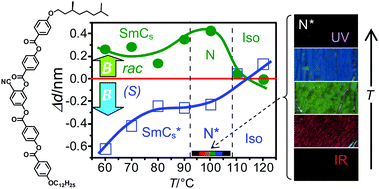
4-Cyanoresorcinol based bent-core compounds combining one branched chiral with one achiral linear end-chain have been synthesized in enatiomerically pure and one compound also in racemic form. All homochiral compounds form a chiral cybotactic nematic phase at relatively low temperature with a selective reflection ranging from the near IR to near UV. For the compound with the longest chains superparaelectric and antiferroelectric switching smectic phases were observed, whereas the corresponding racemate is non-polar. This is attributed to sterically induced polarization by the denser packing of uniform enantiomers due to chirality synchronization of the helical conformers. For the racemic mixture this chirality synchronization requires additional surface stabilization. There are unprecedented effects of an applied magnetic field (1 T) on the LC phases, leading to a layer shrinkage by 6–13% for the enantiomer and a layer expansion by 5–8% for the racemate. It is proposed that the magnetic field couples with the π-systems of the almost rod-like molecules. For the racemate this increases the core order, whereas for the enantiomer the reduction of the heliconical twist is considered to provide the major effect. These magnetic field effects could lead to new applications of chiral LC materials at the cross-over between rod-like and bent shapes.
- This article is part of the themed collection: Editor’s Choice: Advances and New Avenues in Liquid Crystal Science
 Please wait while we load your content...
Please wait while we load your content...

