
A comparative study of Bi, Sb, and BiSb for electrochemical nitrogen reduction leading to a new catalyst design strategy†
 *cd
and
Kyoung-Shin
Choi
*a
*cd
and
Kyoung-Shin
Choi
*a
Abstract
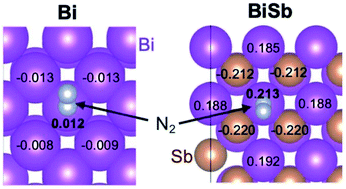
Recent studies identified Bi as one of the most promising non-noble metal elements that can promote the electrochemical N2 reduction reaction (ENRR) to produce NH3. The electronic features that make Bi a promising ENRR catalyst may also be owned by Sb that belongs to the same group as Bi. Thus, the ENRR properties of Bi, Sb, and a BiSb alloy were investigated comparatively to identify common characteristics that facilitate the ENRR. These catalysts were prepared as uniform coating layers on high surface area carbon felt electrodes, which could serve as both regular electrodes and pseudo-gas diffusion electrodes. The experimental results demonstrated that while Bi and Sb show comparable ENRR performances, the formation of a BiSb alloy distinctively increases the faradic efficiency for NH3 production. The X-ray photoelectron spectroscopy results revealed that Bi in BiSb possesses a partial positive charge while Sb in BiSb possesses a partial negative charge, which can impact the way the catalyst surface interacts with the reactants and reaction intermediates of the ENRR and hydrogen evolution reaction (HER), the major competing reaction with the ENRR. Computational investigations including the Bader charge analysis and Gibbs free energy calculations for the elemental steps of the ENRR and HER provided an explanation of how the formation of a BiSb alloy can change the selectivity for the ENRR. The combined experimental and theoretical results and discussion contained in this study lead to a new strategy for designing efficient metal catalysts for the ENRR. Additionally, this study investigated how the use of gas phase and dissolved N2 affected the ENRR performances of the Bi, Sb, and BiSb catalysts.
- This article is part of the themed collection: 2022 Journal of Materials Chemistry Lectureship shortlisted candidates
 Please wait while we load your content...
Please wait while we load your content...

