
C–H and C–F coordination of arenes in neutral alkaline earth metal complexes†

Abstract
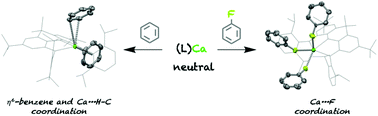
A series of neutral magnesium and calcium complexes bearing an extremely bulky diamido ligand have been synthesised and crystallographically characterised. A number of these complexes feature rare group 2 metal⋯aromatic interactions, such as the η6-coordination of benzene and ‘agostic-like’ C–H coordination, the latter previously unseen in neutral Mg and Ca complexes.
- This article is part of the themed collection: Spotlight Collection: Fluorinated ligands
 Please wait while we load your content...
Please wait while we load your content...

