
β-Catenin/CBP inhibition alters epidermal growth factor receptor fucosylation status in oral squamous cell carcinoma†

Abstract
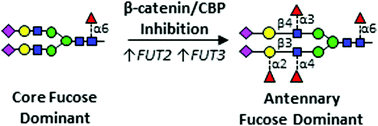
Epidermal growth factor receptor (EGFR) is a major driver of head and neck cancer, a devastating malignancy with a major sub-site in the oral cavity manifesting as oral squamous cell carcinoma (OSCC). EGFR is a glycoprotein receptor tyrosine kinase (RTK) whose activity is upregulated in >80% OSCC. Current anti-EGFR therapy relies on the use of cetuximab, a monoclonal antibody against EGFR, although it has had only a limited response in patients. Here, we uncover a novel mechanism regulating EGFR activity, identifying a role of the nuclear branch of the Wnt/β-catenin signaling pathway, the β-catenin/CBP axis, in control of post-translational modification of N-glycans on the EGFR. Genomic and structural analyses reveal that β-catenin/CBP signaling represses fucosylation on the antennae of N-linked glycans on EGFR. By employing nUPLC-MS/MS, we determined that malignant human OSCC cells harbor EGFR with a paucity of N-glycan antennary fucosylation, while indolent cells display higher levels of fucosylation at sites N420 and N579. Additionally, treatment with either ICG-001 or E7386, which are both small molecule inhibitors of β-catenin/CBP signaling, leads to increased transcriptional expression of fucosyltransferases FUT2 and FUT3, with a concomitant increase in EGFR N-glycan antennary fucosylation. In order to discover which fucosylated glycan epitopes are involved in the observed effect, we performed in-depth characterization of multiply-fucosylated N-glycans via tandem mass spectrometry analysis of the EGFR tryptic glycopeptides. Data are available via ProteomeXchange with identifier PXD017060. We propose that β-catenin/CBP signaling promotes EGFR oncogenic activity in OSCC by inhibiting its N-glycan antennary fucosylation through transcriptional repression of FUT2 and FUT3.
- This article is part of the themed collection: Glycomics & Glycoproteomics: From Analytics to Function
 Please wait while we load your content...
Please wait while we load your content...