
Time-resolved formation of excited atomic and molecular states in XUV-induced nanoplasmas in ammonia clusters†
 *a
Aaron C.
LaForge,
b
Carlo
Callegari,
c
Marcello
Coreno,
f
Michele
Di Fraia,
c
Marcel
Drabbels,
d
Martin
Huppert,
e
Kevin C.
Prince,
c
Stefano
Stranges,
g
Vít
Svoboda,
e
Hans Jakob
Wörner
e
and
Frank
Stienkemeier
a
*a
Aaron C.
LaForge,
b
Carlo
Callegari,
c
Marcello
Coreno,
f
Michele
Di Fraia,
c
Marcel
Drabbels,
d
Martin
Huppert,
e
Kevin C.
Prince,
c
Stefano
Stranges,
g
Vít
Svoboda,
e
Hans Jakob
Wörner
e
and
Frank
Stienkemeier
a
Abstract
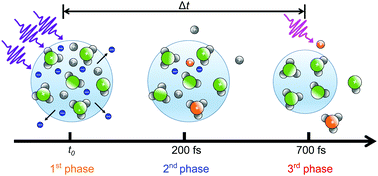
High intensity XUV radiation from a free-electron laser (FEL) was used to create a nanoplasma inside ammonia clusters with the intent of studying the resulting electron–ion interactions and their interplay with plasma evolution. In a plasma-like state, electrons with kinetic energy lower than the local collective Coulomb potential of the positive ionic core are trapped in the cluster and take part in secondary processes (e.g. electron-impact excitation/ionization and electron–ion recombination) which lead to subsequent excited and neutral molecular fragmentation. Using a time-delayed UV laser, the dynamics of the excited atomic and molecular states are probed from −0.1 ps to 18 ps. We identify three different phases of molecular fragmentation that are clearly distinguished by the effect of the probe laser on the ionic and electronic yield. We propose a simple model to rationalize our data and further identify two separate channels leading to the formation of excited hydrogen.
- This article is part of the themed collections: 2020 PCCP HOT Articles and PCCP Editor’s Choice, 2020
 Please wait while we load your content...
Please wait while we load your content...

