
Unexpected monoatomic catalytic-host synergetic OER/ORR by graphitic carbon nitride: density functional theory†
 *a
Changqing
Sun
c
and
*a
Changqing
Sun
c
and
Abstract
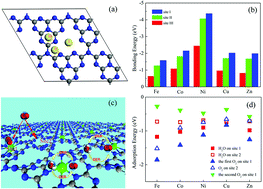
Although single metal atoms (SMAs) have been extensively investigated as unique active sites in single-atom catalysts, the possible active sites of the host catalysts have been unfortunately neglected in previous studies. In single-atom catalysts, the SMAs can promote the chemical and catalytic activities of host atoms, which may act as secondary active sites, resulting in a significant synergistic effect on the catalytic performance. Using density functional theory calculations, we studied the oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) on two different types of active sites: single metal (M1) atoms and the neighboring host atoms of several M1/g-C3N4 samples. The contributions of M1 and host atoms towards the reduction of the OER/ORR overpotentials of Fe1, Co1, Ni1, Cu1 and Zn1/g-C3N4, bifunctional electrocatalysts with the OER/ORR overpotentials of 0.50–0.70 V were investigated. Finally, new M1/g-C3N4 catalysts with high OER/ORR performances could be estimated based on the d-band centre of the M1 atoms in the future.
- This article is part of the themed collection: Editor’s Choice: Computational studies of nanomaterials for energy, catalysis and electronics
 Please wait while we load your content...
Please wait while we load your content...

