
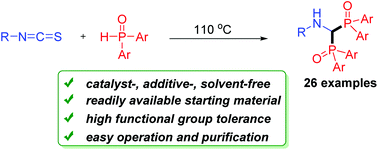
Catalyst- and solvent-free bisphosphinylation of isothiocyanates: a practical method for the synthesis of bisphosphinoylaminomethanes†
Li-Rong
Wen,  ab
Ming
Li
*a
ab
Ming
Li
*a
ab
Ming
Li
*a
Abstract
A general and convenient double addition of phosphine oxides to isothiocyanates is described. It is a practically useful protocol for the construction of bisphosphinoylaminomethanes. The reaction can be carried out smoothly under metal- and solvent-free conditions. It also features a broad substrate scope, simple operation and purification. A possible mechanism involving a tandem double nucleophilic addition/H2S elimination/in situ imine reduction process is proposed.
 Please wait while we load your content...
Please wait while we load your content...

