
Tracking down the origin of peculiar vibrational spectra of aromatic self-assembled thiolate monolayers†
 ab
Shikun
Li,
c
Lyudmila V.
Moskaleva,
c
Armin
Gölzhäuser,
b
Andrey
Turchanin
d
and
Petra
Swiderek
*a
ab
Shikun
Li,
c
Lyudmila V.
Moskaleva,
c
Armin
Gölzhäuser,
b
Andrey
Turchanin
d
and
Petra
Swiderek
*a
Abstract
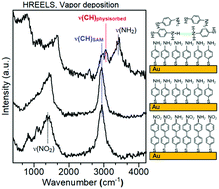
Several studies have previously observed surprisingly low frequencies for the C–H stretching modes of self-assembled monolayers (SAMs) prepared from aromatic thiols. The reason for this property has so far remained elusive. Therefore, we report a novel study of the vibrational spectra of SAMs prepared on Au from two different aromatic thiols, namely, 4′-nitro-1,1′-biphenyl-4-thiol (NBPT) and 4-aminothiophenol (ATP). The SAMs were prepared by vapor deposition (VD) in ultrahigh vacuum (UHV) as well as by the solution method (SM) and their quality was controlled by X-ray photoelectron spectroscopy (XPS). In addition, amino terminated SAMs were also obtained by electron irradiation and by chemical reduction of NBPT SAMs. Beside infrared reflection absorption spectroscopy (IRRAS), we have employed high resolution electron energy loss spectroscopy (HREELS), by which VD SAMs can be studied in situ, i.e. without exposing them to air. Hence, we can exclude possible contributions of solvent molecules to the vibrational spectra. Nonetheless, HREELS in fact reveals the same large red shift of the C–H stretching modes in the SAMs as also observed in ex situ IRRAS experiments. In contrast, HREELS for physisorbed ATP and ATP in a KBr pellet measured by transmission infrared spectroscopy exhibit the expected aromatic bands. Using a computational approach, we can exclude molecular packing effects as origin of this shift. Therefore, we propose chemical changes in the aromatic rings during SAM formation as an alternative explanation for the observed frequency shift. As another striking effect, the N–H stretching vibrational modes of the amino-terminated SAMs are extremely weak in both IRRAS and HREELS despite the fact that XPS confirms the presence of amino groups. A very weak signal is observed only in the case of an electron irradiated NBPT SAM. In contrast, an energy loss ascribed to the N–H stretching vibrations is clearly observed in HREELS of ATP physisorbed on an ATP SAM and on graphite as well as in the transmission infrared spectrum of ATP in KBr. The extremely low intensity of these vibrations in the SAM is traced back to the inherently low transition dipole moment for the excitation of N–H stretching modes in free N–H groups. Furthermore, the calculations suggest that the much stronger signals of N–H stretching modes involved in hydrogen-bonding with adjacent amino groups are suppressed because these vibrations are oriented parallel to the surface.
 Please wait while we load your content...
Please wait while we load your content...

