
Self-aggregation and coaggregation of the p53 core fragment with its aggregation gatekeeper variant†
Abstract
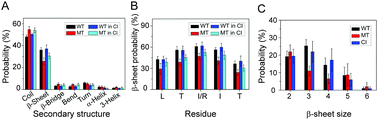
Recent studies suggested that p53 aggregation can lead to loss-of-function (LoF), dominant-negative (DN) and gain-of-function (GoF) effects, with adverse cancer consequences. The p53 aggregation-nucleating 251ILTIITL257 fragment is a key segment in wild-type p53 aggregation; however, an I254R mutation can prevent it. It was suggested that self-assembly of wild-type p53 and its cross-interaction with mutants differ from the classical amyloid nucleation-growth mechanism. Here, using replica exchange molecular dynamics (REMD) simulations, we studied the cross-interactions of this p53 core fragment and its aggregation rescue I254R mutant. We found that the core fragment displays strong aggregation propensity, whereas the gatekeeper I254R mutant tends to be disordered, consistent with experiments. Our cross-interaction results reveal that the wild-type p53 fragment promotes β-sheet formation of the I254R mutant by shifting the disordered mutant peptides into aggregating states. As a result, the system has similar oligomeric structures, inter-peptide interactions and free energy landscape as the wild type fragment does, revealing a prion-like process. We also found that in the cross-interaction system, the wild-type species has higher tendency to interact with the mutant than with itself. This phenomenon illustrates synergistic effects between the p53 251ILTIITL257 fragment and the mutant resembling prion cross-species propagation, cautioning against exploiting it in drug discovery.
 Please wait while we load your content...
Please wait while we load your content...

