
Vibrational spectroscopy of Cu+(H2)4: about anharmonicity and fluxionality†

Abstract
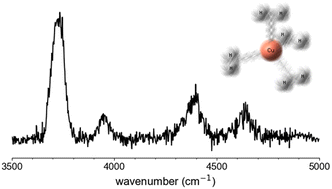
The vibrational spectra of the copper(I) cation–dihydrogen complexes Cu+(H2)4, Cu+(D2)4 and Cu+(D2)3H2 are studied using cryogenic ion trap vibrational spectroscopy in combination with quantum chemical calculations. The infrared photodissociation (IRPD) spectra (2500–7300 cm−1) are assigned based on a comparison to IR spectra calculated using vibrational second-order perturbation theory (VPT2). The IRPD spectra exhibit ≈60 cm−1 broad bands that lack rotational resolution, indicative of rather floppy complexes even at an ion trap temperature of 10 K. The observed vibrational features are assigned to the excitations of dihydrogen stretching fundamentals, combination bands of these fundamentals with low energy excitations as well as overtone excitations of a minimum-energy structure with Cs symmetry. The three distinct dihydrogen positions present in the structure can interconvert via pseudorotations with energy barriers less than 10 cm−1, far below the zero-point vibrational energy. Ab initio Born–Oppenheimer molecular dynamics (BOMD) simulations confirm the fluxional behavior of these complexes and yield an upper limit for the timeframe of the pseudorotation on the order of 10 ps. For Cu+(D2)3H2, the H2 and D2 loss channels yield different IRPD spectra indicating non-ergodic behavior.
 Please wait while we load your content...
Please wait while we load your content...

