
Modelling amorphous materials via a joint solid-state NMR and X-ray absorption spectroscopy and DFT approach: application to alumina†
 a
Steffen P.
Emge,
b
Pieter C. M. M.
Magusin,
bc
Clare P.
Grey
b
and
Andrew J.
Morris
*d
a
Steffen P.
Emge,
b
Pieter C. M. M.
Magusin,
bc
Clare P.
Grey
b
and
Andrew J.
Morris
*d
Abstract
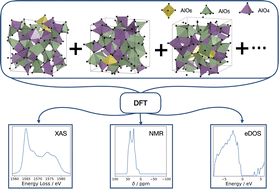
Understanding a material's electronic structure is crucial to the development of many functional devices from semiconductors to solar cells and Li-ion batteries. A material's properties, including electronic structure, are dependent on the arrangement of its atoms. However, structure determination (the process of uncovering the atomic arrangement), is impeded, both experimentally and computationally, by disorder. The lack of a verifiable atomic model presents a huge challenge when designing functional amorphous materials. Such materials may be characterised through their local atomic environments using, for example, solid-state NMR and XAS. By using these two spectroscopy methods to inform the sampling of configurations from ab initio molecular dynamics we devise and validate an amorphous model, choosing amorphous alumina to illustrate the approach due to its wide range of technological uses. Our model predicts two distinct geometric environments of AlO5 coordination polyhedra and determines the origin of the pre-edge features in the Al K-edge XAS. From our model we construct an average electronic density of states for amorphous alumina, and identify localized states at the conduction band minimum (CBM). We show that the presence of a pre-edge peak in the XAS is a result of transitions from the Al 1s to Al 3s states at the CBM. Deconvoluting this XAS by coordination geometry reveals contributions from both AlO4 and AlO5 geometries at the CBM give rise to the pre-edge, which provides insight into the role of AlO5 in the electronic structure of alumina. This work represents an important advance within the field of solid-state amorphous modelling, providing a method for developing amorphous models through the comparison of experimental and computationally derived spectra, which may then be used to determine the electronic structure of amorphous materials.
 Please wait while we load your content...
Please wait while we load your content...

