
Active learning of chemical reaction networks via probabilistic graphical models and Boolean reaction circuits†

Abstract
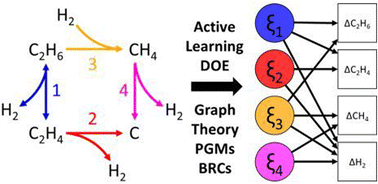
Discerning networks of many reactions among multiple interconverting species is challenging. Here, we present a reaction network identification methodology. Our methodology enumerates all stoichiometrically and chemically feasible reactions and requires statistical evidence from effluent concentrations for the inclusion or exclusion of each from the reaction network, contrasting with the commonly seen incremental approach and other work of relying heavily upon chemical intuition and assuming the reactions occurring. Using graph theory alongside an active learning design of experiments that propose maximally informative feeds, we identify the underlying reaction network with minimal laboratory runs. We introduce chemistry-probabilistic graphical modeling and Boolean reaction circuits to statistically quantify which reactions occur from effluent concentrations. Our methodology accurately discerns active reactions, as showcased upon a laboratory network of cross-ketonization of furoic and lauric acid and validated upon simulated networks of thermal and CO2-assisted ethane dehydrogenation.
 Please wait while we load your content...
Please wait while we load your content...

