
MolE8: finding DFT potential energy surface minima values from force-field optimised organic molecules with new machine learning representations†

Abstract
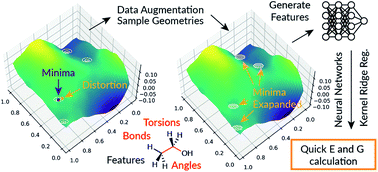
The use of machine learning techniques in computational chemistry has gained significant momentum since large molecular databases are now readily available. Predictions of molecular properties using machine learning have advantages over the traditional quantum mechanics calculations because they can be cheaper computationally without losing the accuracy. We present a new extrapolatable and explainable molecular representation based on bonds, angles and dihedrals that can be used to train machine learning models. The trained models can accurately predict the electronic energy and the free energy of small organic molecules with atom types C, H N and O, with a mean absolute error of 1.2 kcal mol−1. The models can be extrapolated to larger organic molecules with an average error of less than 3.7 kcal mol−1 for 10 or fewer heavy atoms, which represent a chemical space two orders of magnitude larger. The rapid energy predictions of multiple molecules, up to 7 times faster than previous ML models of similar accuracy, has been achieved by sampling geometries around the potential energy surface minima. Therefore, the input geometries do not have to be located precisely on the minima and we show that accurate density functional theory energy predictions can be made from force-field optimised geometries with a mean absolute error 2.5 kcal mol−1.
 Please wait while we load your content...
Please wait while we load your content...

