
Computationally driven discovery of SARS-CoV-2 Mpro inhibitors: from design to experimental validation†‡
 §a
Zhifeng
Jing,
§a
Florent
Hédin,
a
Dina
El Ahdab,
n
Cristiano
Salata,
h
Stefano
Moro,
g
Jay W.
Ponder,
lm
Jean-Philip
Piquemal
*no
and
§a
Zhifeng
Jing,
§a
Florent
Hédin,
a
Dina
El Ahdab,
n
Cristiano
Salata,
h
Stefano
Moro,
g
Jay W.
Ponder,
lm
Jean-Philip
Piquemal
*no
and
Abstract
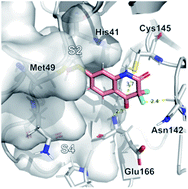
We report a fast-track computationally driven discovery of new SARS-CoV-2 main protease (Mpro) inhibitors whose potency ranges from mM for the initial non-covalent ligands to sub-μM for the final covalent compound (IC50 = 830 ± 50 nM). The project extensively relied on high-resolution all-atom molecular dynamics simulations and absolute binding free energy calculations performed using the polarizable AMOEBA force field. The study is complemented by extensive adaptive sampling simulations that are used to rationalize the different ligand binding poses through the explicit reconstruction of the ligand–protein conformation space. Machine learning predictions are also performed to predict selected compound properties. While simulations extensively use high performance computing to strongly reduce the time-to-solution, they were systematically coupled to nuclear magnetic resonance experiments to drive synthesis and for in vitro characterization of compounds. Such a study highlights the power of in silico strategies that rely on structure-based approaches for drug design and allows the protein conformational multiplicity problem to be addressed. The proposed fluorinated tetrahydroquinolines open routes for further optimization of Mpro inhibitors towards low nM affinities.
 Please wait while we load your content...
Please wait while we load your content...

