
Revealing initial nucleation of hexagonal boron nitride on Ru(0001) and Rh(111) surfaces by density functional theory simulations
 *a
*a
Abstract
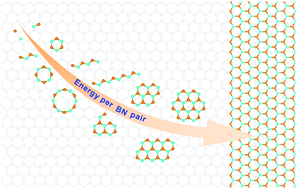
Hexagonal boron nitride (h-BN) has attracted extensive attention due to its broad applications in electronic devices. Among various methods, the chemical vapor deposition (CVD) method has the most potential for obtaining high-quality h-BN films. A deep understanding of the initial nucleation process is essential for the rationally designed synthesis of h-BN. In this work, we systematically studied the stabilities of various BN clusters on Ru(0001) and Rh(111) surfaces by density functional theory simulations. Our results show that geometry types, docking sites, edged atoms, symmetry of geometries, and metal substrates can influence the stabilities of BN clusters. A structure crossover from a chain-like to the sp2 honeycomb geometry occurs at a critical size of 6 on both metal surfaces. After this, the honeycomb geometry is the energy-favoured one and down-hill growth continues until full coverage of h-BN is reached. The nucleation of h-BN is an energy-driven process governed by the charge transfer between BN clusters and underlying metals. The atomic scale understanding should provide helpful information on the rationally designed synthesis of h-BN and other binary 2D materials.
 Please wait while we load your content...
Please wait while we load your content...

