
A mechanistic study on the gold(i)-catalyzed cyclization of propargylic amide: revealing the impact of expanded-ring N-heterocyclic carbenes†

Abstract
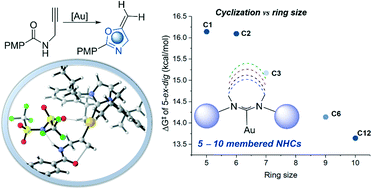
The interest in expanded-ring N-heterocyclic carbenes (ER-NHCs) has recently received much attention, especially with the Au(I)-catalyzed activation of alkynes. Herein, we report density functional theory (DFT) investigations on the Au(I)-catalyzed cyclization of propargylic amides to exploit the mechanistic effect of variable ER-NHCs to shed some light for further future developments. Mechanistically, the reaction undergoes a stepwise intramolecular nucleophilic addition after the π-complexation step with the alkyne moiety, while the counteranion interacts with the amide group. Subsequently, the N-deprotonation followed by C-protonation (protodeauration) process furnishes the cyclized product, and regenerates the LAuNTf2 to continue the catalytic cycle. Although the deprotonation–protonation process enabled by the counteranion (NTf2−) is slow, it is significantly promoted by the oxazole product. Thus, the reaction is suggested to be autocatalyzed. Both cyclization and protonation steps favor the 5-exo over 6-endo product with unsubstituted terminal alkyne. The ring-size effect of NHCs is explored, where NHCs larger than the 5-membered ring provide intrinsically larger steric demand with the same aryl group on it, which is shown to inhibit the reactivity. For NHCs with similar steric properties, ER-NHCs accelerate the cyclization step. Various electronic structure analyses show that for the Au(I) center, ER-NHCs are less effective electron donors because of less orbital overlap and render the Au(I) more electrophilic. This work provides new dimensions to the development of Au(I)-catalyzed methodologies to engineering ligands.
 Please wait while we load your content...
Please wait while we load your content...

