
Auto-QChem: an automated workflow for the generation and storage of DFT calculations for organic molecules†

Abstract
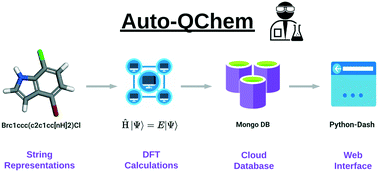
This perspective describes Auto-QChem, an automatic, high-throughput and end-to-end DFT calculation workflow that computes chemical descriptors for organic molecules. Tailored toward users without extensive programming experience, Auto-QChem has facilitated more than 38 000 DFT calculations for 17 000 molecules as of January 2022. Starting from string representations of molecules, Auto-QChem automatically (a) generates conformational ensembles, (b) submits and manages DFT calculations on a high-performance computing (HPC) cluster, (c) extracts production-ready features that are suitable for statistical analysis and machine learning model development, and (d) stores resulting calculations in a cloud-hosted and web-accessible database. We describe in detail the design and implementation of Auto-QChem, as well as its current functionalities. We also review three case studies where Auto-QChem was applied to our recent efforts in combining data science approaches in organic chemistry methodology development: (a) the design of a diverse and unbiased aryl bromide substrate scope for a Ni/photoredox catalyzed alkylation reaction, (b) mechanistic studies on the effect of bioxazoline (BiOx) and biimidazoline (BiIm) ligands on enantioselectivity in a Ni/photoredox catalyzed cross-electrophile coupling of epoxides and aryl iodides, (c) the development of a reaction condition optimization framework using Bayesian optimization. In addition, we discuss limitations and future directions of Auto-QChem and similar automated DFT calculation systems.
 Please wait while we load your content...
Please wait while we load your content...

