
Long-range dispersion-inclusive machine learning potentials for structure search and optimization of hybrid organic–inorganic interfaces†
 a
Shayantan
Chaudhuri,
ab
Andreas
Jeindl,
c
Oliver T.
Hofmann
c
and
Reinhard J.
Maurer
*a
a
Shayantan
Chaudhuri,
ab
Andreas
Jeindl,
c
Oliver T.
Hofmann
c
and
Reinhard J.
Maurer
*a
Abstract
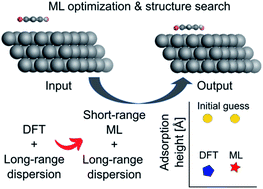
The computational prediction of the structure and stability of hybrid organic–inorganic interfaces provides important insights into the measurable properties of electronic thin film devices, coatings, and catalyst surfaces and plays an important role in their rational design. However, the rich diversity of molecular configurations and the important role of long-range interactions in such systems make it difficult to use machine learning (ML) potentials to facilitate structure exploration that otherwise requires computationally expensive electronic structure calculations. We present an ML approach that enables fast, yet accurate, structure optimizations by combining two different types of deep neural networks trained on high-level electronic structure data. The first model is a short-ranged interatomic ML potential trained on local energies and forces, while the second is an ML model of effective atomic volumes derived from atoms-in-molecules partitioning. The latter can be used to connect short-range potentials to well-established density-dependent long-range dispersion correction methods. For two systems, specifically gold nanoclusters on diamond (110) surfaces and organic π-conjugated molecules on silver (111) surfaces, we train models on sparse structure relaxation data from density functional theory and show the ability of the models to deliver highly efficient structure optimizations and semi-quantitative energy predictions of adsorption structures.
- This article is part of the themed collections: #RSCPoster Conference, Celebrating our 2024 Prizewinners and Machine Learning and Artificial Intelligence: A cross-journal collection
 Please wait while we load your content...
Please wait while we load your content...

