
Ca4Sb2O and Ca4Bi2O: two promising mixed-anion thermoelectrics†

Abstract
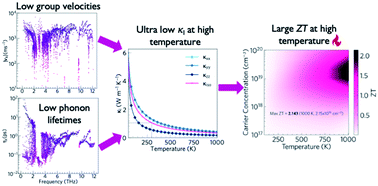
The environmental burden of fossil fuels and the rising impact of global warming have created an urgent need for sustainable clean energy sources. This has led to widespread interest in thermoelectric (TE) materials to recover part of the ∼60% of global energy currently wasted as heat as usable electricity. Oxides are particularly attractive as they are thermally stable, chemically inert, and formed of earth-abundant elements, but despite intensive efforts there have been no reports of oxide TEs matching the performance of flagship chalcogenide materials such as PbTe, Bi2Te3 and SnSe. A number of ternary X4Y2Z mixed-anion systems, including oxides, have predicted band gaps in the useful range for several renewable-energy applications, including as TEs, and some also show the complex crystal structures indicative of low lattice thermal conductivity. In this study, we use ab initio calculations to investigate the TE performance of two structurally-similar mixed-anion oxypnictides, Ca4Sb2O and Ca4Bi2O. Electronic-structure and band-alignment calculations using hybrid density-functional theory (DFT), including spin–orbit coupling, suggest that both materials are likely to be p-type dopable with large charge-carrier mobilities. Lattice-dynamics calculations using third-order perturbation theory predict ultra-low lattice thermal conductivities of ∼0.8 and ∼0.5 W m−1 K−1 above 750 K. Nanostructuring to a crystal grain size of 20 nm is predicted to further reduce the room temperature thermal conductivity by around 40%. Finally, we use the electronic- and thermal-transport calculations to estimate the thermoelectric figure of merit ZT, and show that with p-type doping both oxides could potentially serve as promising earth-abundant oxide TEs for high-temperature applications.
 Please wait while we load your content...
Please wait while we load your content...

