Structural transitions at electrodes, immersed in simple ionic liquid models

Abstract
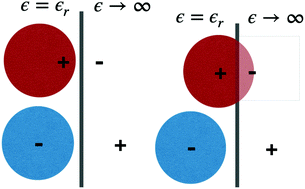
We used a recently developed classical Density Functional Theory (DFT) method to study the structures, phase transitions, and electrochemical behaviours of two coarse-grained ionic fluid models, in the presence of a perfectly conducting model electrode. Common to both is that the charge of the cationic component is able to approach the electrode interface more closely than the anion charge. This means that the cations are specifically attracted to the electrode, due to surface polarization effects. Hence, for a positively charged electrode, there is competition at the surface between cations and anions, where the latter are attracted by the positive electrode charge. This generates demixing, for a range of positive voltages, where the two phases are structurally quite different. The surface charge density is also different between the two phases, even at the same potential. The DFT formulation contains an approximate treatment of ion correlations, and surface polarization, where the latter is modelled via screened image interactions. Using a mean-field DFT, where ion correlations are neglected, causes the phase transition to vanish for both models, but there is still a dramatic drop in the differential capacitance as proximal cations are replaced by anions, for increasing surface potentials. While these findings were obtained for relatively crude coarse-grained models, we argue that the findings can also be relevant in “real” systems, where we note that many ionic liquids are composed of a spherically symmetric anion, and a cation that is asymmetric both from a steric and a charge distribution point of view.
 Please wait while we load your content...
Please wait while we load your content...

