
Inhibitor binding influences the protonation states of histidines in SARS-CoV-2 main protease†
 a
Isabella
Daidone,
b
Laura
Zanetti-Polzi,
c
Daniel W.
Kneller,
g
Andrey
Kovalevsky,
g
Leighton
Coates,
g
Josh V.
Vermaas,
i
Yui Tik
Pang,
a
Atanu
Acharya,
a
Jerry M.
Parks,
j
James C.
Gumbart
*a
a
Isabella
Daidone,
b
Laura
Zanetti-Polzi,
c
Daniel W.
Kneller,
g
Andrey
Kovalevsky,
g
Leighton
Coates,
g
Josh V.
Vermaas,
i
Yui Tik
Pang,
a
Atanu
Acharya,
a
Jerry M.
Parks,
j
James C.
Gumbart
*a
Abstract
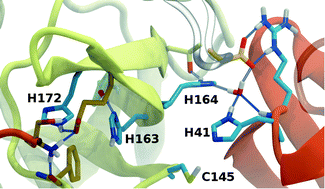
The main protease (Mpro) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is an attractive target for antiviral therapeutics. Recently, many high-resolution apo and inhibitor-bound structures of Mpro, a cysteine protease, have been determined, facilitating structure-based drug design. Mpro plays a central role in the viral life cycle by catalyzing the cleavage of SARS-CoV-2 polyproteins. In addition to the catalytic dyad His41–Cys145, Mpro contains multiple histidines including His163, His164, and His172. The protonation states of these histidines and the catalytic nucleophile Cys145 have been debated in previous studies of SARS-CoV Mpro, but have yet to be investigated for SARS-CoV-2. In this work we have used molecular dynamics simulations to determine the structural stability of SARS-CoV-2 Mpro as a function of the protonation assignments for these residues. We simulated both the apo and inhibitor-bound enzyme and found that the conformational stability of the binding site, bound inhibitors, and the hydrogen bond networks of Mpro are highly sensitive to these assignments. Additionally, the two inhibitors studied, the peptidomimetic N3 and an α-ketoamide, display distinct His41/His164 protonation-state-dependent stabilities. While the apo and the N3-bound systems favored Nδ (HD) and Nϵ (HE) protonation of His41 and His164, respectively, the α-ketoamide was not stably bound in this state. Our results illustrate the importance of using appropriate histidine protonation states to accurately model the structure and dynamics of SARS-CoV-2 Mpro in both the apo and inhibitor-bound states, a necessary prerequisite for drug-design efforts.
- This article is part of the themed collections: Coronavirus articles - free to access collection and Most popular 2021 physical and theoretical chemistry articles, 2021
 Please wait while we load your content...
Please wait while we load your content...

