
Theoretical studies on the initial reaction kinetics and mechanisms of p-, m- and o-nitrotoluene†
 a
Peng
Zhang,
b
a
Peng
Zhang,
b
Abstract
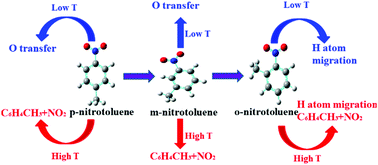
The potential energy surfaces (PESs) of three nitrotoluene isomers, such as p-nitrotoluene, m-nitrotoluene, and o-nitrotoluene, have been theoretically built at the CCSD(T)/CBS level. The geometries of reactants, transition states (TSs) and products are optimized at the B3LYP/6-311++G(d,p) level. Results show that reactions of –NO2 isomerizing to ONO, and C–NO2 bond dissociation play important roles among all of the initial channels for p-nitrotoluene and m-nitrotoluene, and that the H atom migration and C–NO2 bond dissociation are dominant reactions for o-nitrotoluene. In addition, there exist pathways for three isomer conversions, but with high energy barriers. Rate constant calculations and branching ratio analyses further demonstrate that the isomerization reactions of O transfer are prominent at low to intermediate temperatures, whereas the direct C–NO2 bond dissociation reactions prevail at high temperatures for p-nitrotoluene and m-nitrotoluene, and that H atom migration is a predominant reaction for o-nitrotoluene, while C–NO2 bond dissociation becomes important by increasing the temperature.
 Please wait while we load your content...
Please wait while we load your content...

