
Reaction mechanisms of C(3PJ) and C+(2PJ) with benzene in the interstellar medium from quantum mechanical molecular dynamics simulations†

Abstract
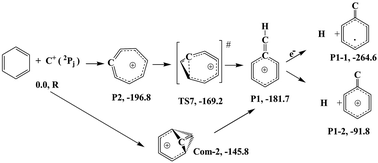
While spectroscopic data on small hydrocarbons in interstellar media in combination with crossed molecular beam (CMB) experiments have provided a wealth of information on astrochemically relevant species, much of the underlying mechanistic pathways of their formation remain elusive. Therefore, in this work, the chemical reaction mechanisms of C(3PJ) + C6H6 and C+(2P) + C6H6 systems using the quantum mechanical molecular dynamics (QMMD) technique at the PBE0-D3(BJ) level of theory is investigated, mimicking a CMB experiment. Both the dynamics of the reactions as well as the electronic structure for the purpose of the reaction network are evaluated. The method is validated for the first reaction by comparison to the available experimental data. The reaction scheme for the C(3PJ) + C6H6 system covers the literature data, e.g. the major products are the 1,2-didehydrocycloheptatrienyl radical (C7H5) and benzocyclopropenyl radical (C6H5–CH), and it reveals the existence of less common pathways for the first time. The chemistry of the C+(2PJ) + C6H6 system is found to be much richer, and we have found that this is because of more exothermic reactions in this system in comparison to those in the C(3PJ) + C6H6 system. Moreover, using the QMMD simulation, a number of reaction paths have been revealed that produce three distinct classes of reaction products with different ring sizes. All in all, at all the collision energies and orientations, the major product is the heptagon molecular ion for the ionic system. It is also revealed that the collision orientation has a dominant effect on the reaction products in both systems, while the collision energy mostly affects the charged system. These simulations both prove the applicability of this approach to simulate crossed molecular beams, and provide fundamental information on reactions relevant for the interstellar medium.
 Please wait while we load your content...
Please wait while we load your content...

