
The local atomic structure and thermoelectric properties of Ir-doped ZnO: hybrid DFT calculations and XAS experiments†
 a
Denis
Gryaznov,
*a
Andrea
Zitolo,
e
Martins
Zubkins,
a
Alexei
Kuzmin,
a
Andris
Anspoks
a
and
a
Denis
Gryaznov,
*a
Andrea
Zitolo,
e
Martins
Zubkins,
a
Alexei
Kuzmin,
a
Andris
Anspoks
a
and
Abstract
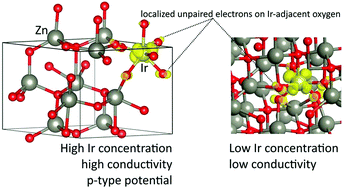
We combined the hybrid density functional theory (DFT) calculations and X-ray absorption spectroscopy (XAS) experiments in the study of the local atomic structure around Ir ions in ZnO thin films with different iridium content. This was then used in the first principles analysis of the thermoelectric properties of material. The emphasis has been put on the conditions for a positive Seebeck coefficient and p-type electrical conductivity as the functions of the Fermi level. We studied both computationally and experimentally several possible IrOx polyhedra (complexes) with a different number of surrounding oxygens and Ir oxidation states, including those with the formation of peroxide ions (O22−). In particular, octahedral coordination of iridium ions was identified by reverse Monte Carlo (RMC) simulations of the Ir L3-edge EXAFS spectra of ZnO:Ir thin films as the predominant complex, which is supported by the calculated lowest interstitial oxygen incorporation energies. All the calculated IrOx (x = 4, 5, 6) complexes, regardless of Ir the oxidation state, demonstrate potential for p-type conduction if the Fermi level (μF) falls in the range of 0–0.8 eV from the valence band maximum (VBM) and the Ir concentration is high enough (12.5% in the present DFT calculations). Even though the corresponding calculated Seebeck coefficient (S) around 80–89 μV K−1 slightly exceeds the experimental values, we emphasise the presence of an important plateau in the dependence of S on μF in this range for two complexes with the formation of peroxide ions (O22−). We predicted also that peroxide ions O22− are characterized by the calculated phonon frequencies of 810–942 cm−1 in agreement with our previous Raman experimental results. In this light, we discuss the high sensitivity of calculated S(μF) dependences to the atomic and electronic structure.
 Please wait while we load your content...
Please wait while we load your content...

