
Real-time prediction of 1H and 13C chemical shifts with DFT accuracy using a 3D graph neural network†
 *a
S. V.
Shree Sowndarya,
a
Liliana C.
Gallegos,
a
Peter C.
St. John
b
and
Robert S.
Paton
*a
*a
S. V.
Shree Sowndarya,
a
Liliana C.
Gallegos,
a
Peter C.
St. John
b
and
Robert S.
Paton
*a
Abstract
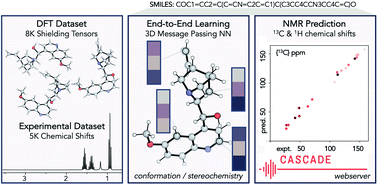
Nuclear magnetic resonance (NMR) is one of the primary techniques used to elucidate the chemical structure, bonding, stereochemistry, and conformation of organic compounds. The distinct chemical shifts in an NMR spectrum depend upon each atom's local chemical environment and are influenced by both through-bond and through-space interactions with other atoms and functional groups. The in silico prediction of NMR chemical shifts using quantum mechanical (QM) calculations is now commonplace in aiding organic structural assignment since spectra can be computed for several candidate structures and then compared with experimental values to find the best possible match. However, the computational demands of calculating multiple structural- and stereo-isomers, each of which may typically exist as an ensemble of rapidly-interconverting conformations, are expensive. Additionally, the QM predictions themselves may lack sufficient accuracy to identify a correct structure. In this work, we address both of these shortcomings by developing a rapid machine learning (ML) protocol to predict 1H and 13C chemical shifts through an efficient graph neural network (GNN) using 3D structures as input. Transfer learning with experimental data is used to improve the final prediction accuracy of a model trained using QM calculations. When tested on the CHESHIRE dataset, the proposed model predicts observed 13C chemical shifts with comparable accuracy to the best-performing DFT functionals (1.5 ppm) in around 1/6000 of the CPU time. An automated prediction webserver and graphical interface are accessible online at http://nova.chem.colostate.edu/cascade/. We further demonstrate the model in three applications: first, we use the model to decide the correct organic structure from candidates through experimental spectra, including complex stereoisomers; second, we automatically detect and revise incorrect chemical shift assignments in a popular NMR database, the NMRShiftDB; and third, we use NMR chemical shifts as descriptors for determination of the sites of electrophilic aromatic substitution.
- This article is part of the themed collection: Most popular 2021 physical and theoretical chemistry articles, 2021
 Please wait while we load your content...
Please wait while we load your content...

