
Rapid and accurate estimation of protein–ligand relative binding affinities using site-identification by ligand competitive saturation†
 ‡a
Anthony
Hazel,
‡a
Vincent D.
Ustach,
a
Sunhwan
Jo,
b
Alexander D.
MacKerell, Jr
*ab
‡a
Anthony
Hazel,
‡a
Vincent D.
Ustach,
a
Sunhwan
Jo,
b
Alexander D.
MacKerell, Jr
*ab
Abstract
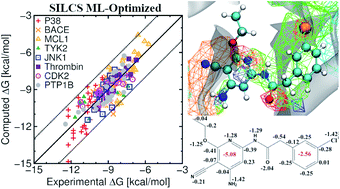
Predicting relative protein–ligand binding affinities is a central pillar of lead optimization efforts in structure-based drug design. The site identification by ligand competitive saturation (SILCS) methodology is based on functional group affinity patterns in the form of free energy maps that may be used to compute protein–ligand binding poses and affinities. Presented are results obtained from the SILCS methodology for a set of eight target proteins as reported originally in Wang et al. (J. Am. Chem. Soc., 2015, 137, 2695–2703) using free energy perturbation (FEP) methods in conjunction with enhanced sampling and cycle closure corrections. These eight targets have been subsequently studied by many other authors to compare the efficacy of their method while comparing with the outcomes of Wang et al. In this work, we present results for a total of 407 ligands on the eight targets and include specific analysis on the subset of 199 ligands considered previously. Using the SILCS methodology we can achieve an average accuracy of up to 77% and 74% when considering the eight targets with their 199 and 407 ligands, respectively, for rank-ordering ligand affinities as calculated by the percent correct metric. This accuracy increases to 82% and 80%, respectively, when the SILCS atomic free energy contributions are optimized using a Bayesian Markov-chain Monte Carlo approach. We also report other metrics including Pearson's correlation coefficient, Pearlman's predictive index, mean unsigned error, and root mean square error for both sets of ligands. The results obtained for the 199 ligands are compared with the outcomes of Wang et al. and other published works. Overall, the SILCS methodology yields similar or better-quality predictions without a priori need for known ligand orientations in terms of the different metrics when compared to current FEP approaches with significant computational savings while additionally offering quantitative estimates of individual atomic contributions to binding free energies. These results further validate the SILCS methodology as an accurate, computationally efficient tool to support lead optimization and drug discovery.
 Please wait while we load your content...
Please wait while we load your content...

