
2-Mercaptomethyl-thiazolidines use conserved aromatic–S interactions to achieve broad-range inhibition of metallo-β-lactamases†
 ‡a
Veronica
Martinez,
‡b
Philip
Hinchliffe,
‡c
Maria F.
Mojica,
def
Diego M.
Moreno,
gj
James
Spencer,
c
Alejandro J.
Vila
*aj
and
Graciela
Mahler
*b
‡a
Veronica
Martinez,
‡b
Philip
Hinchliffe,
‡c
Maria F.
Mojica,
def
Diego M.
Moreno,
gj
James
Spencer,
c
Alejandro J.
Vila
*aj
and
Graciela
Mahler
*b
Abstract
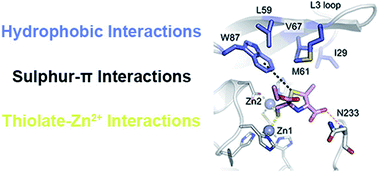
Infections caused by multidrug resistant (MDR) bacteria are a major public health threat. Carbapenems are among the most potent antimicrobial agents that are commercially available to treat MDR bacteria. Bacterial production of carbapenem-hydrolysing metallo-β-lactamases (MBLs) challenges their safety and efficacy, with subclass B1 MBLs hydrolysing almost all β-lactam antibiotics. MBL inhibitors would fulfil an urgent clinical need by prolonging the lifetime of these life-saving drugs. Here we report the synthesis and activity of a series of 2-mercaptomethyl-thiazolidines (MMTZs), designed to replicate MBL interactions with reaction intermediates or hydrolysis products. MMTZs are potent competitive inhibitors of B1 MBLs in vitro (e.g., Ki = 0.44 μM vs. NDM-1). Crystal structures of MMTZ complexes reveal similar binding patterns to the most clinically important B1 MBLs (NDM-1, VIM-2 and IMP-1), contrasting with previously studied thiol-based MBL inhibitors, such as bisthiazolidines (BTZs) or captopril stereoisomers, which exhibit lower, more variable potencies and multiple binding modes. MMTZ binding involves thiol coordination to the Zn(II) site and extensive hydrophobic interactions, burying the inhibitor more deeply within the active site than D/L-captopril. Unexpectedly, MMTZ binding features a thioether–π interaction with a conserved active-site aromatic residue, consistent with their equipotent inhibition and similar binding to multiple MBLs. MMTZs penetrate multiple Enterobacterales, inhibit NDM-1 in situ, and restore carbapenem potency against clinical isolates expressing B1 MBLs. Based on their inhibitory profile and lack of eukaryotic cell toxicity, MMTZs represent a promising scaffold for MBL inhibitor development. These results also suggest sulphur–π interactions can be exploited for general ligand design in medicinal chemistry.
 Please wait while we load your content...
Please wait while we load your content...

