
Data-driven coarse-grained modeling of polymers in solution with structural and dynamic properties conserved

Abstract
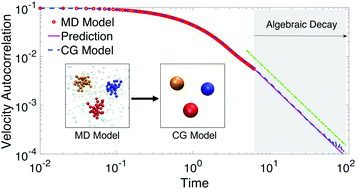
We present data-driven coarse-grained (CG) modeling for polymers in solution, which conserves the dynamic as well as structural properties of the underlying atomistic system. The CG modeling is built upon the framework of the generalized Langevin equation (GLE). The key is to determine each term in the GLE by directly linking it to atomistic data. In particular, we propose a two-stage Gaussian process-based Bayesian optimization method to infer the non-Markovian memory kernel from the data of the velocity autocorrelation function (VACF). Considering that the long-time behaviors of the VACF and memory kernel for polymer solutions can exhibit hydrodynamic scaling (algebraic decay with time), we further develop an active learning method to determine the emergence of hydrodynamic scaling, which can accelerate the inference process of the memory kernel. The proposed methods do not rely on how the mean force or CG potential in the GLE is constructed. Thus, we also compare two methods for constructing the CG potential: a deep learning method and the iterative Boltzmann inversion method. With the memory kernel and CG potential determined, the GLE is mapped onto an extended Markovian process to circumvent the expensive cost of directly solving the GLE. The accuracy and computational efficiency of the proposed CG modeling are assessed in a model star-polymer solution system at three representative concentrations. By comparing with the reference atomistic simulation results, we demonstrate that the proposed CG modeling can robustly and accurately reproduce the dynamic and structural properties of polymers in solution.
 Please wait while we load your content...
Please wait while we load your content...

