
The origin of regioselectivity in Cu-catalyzed hydrocarbonylative coupling of alkynes with alkyl halides†

Abstract
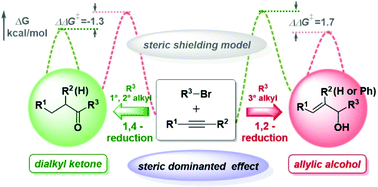
In recent years, the versatile reactivity of Cu-catalyzed hydrocarbonylative coupling of alkynes with alkyl halides has drawn widespread attention. In this paper, we explore in detail the origin of different regioselectivities for terminal/internal alkynes coupling with primary, secondary and tertiary alkyl halides by using density functional theory (DFT) calculations. The present results reveal that the dominant factor of high Cα-regioselectivity in alkyne insertion is mainly the electron effect for the terminal alkyne, in which the higher electron density of terminal carbon makes electrophilic attack more favorable. For internal alkynes, such as 1-phenyl-1-hexyne, the d orbital in Cu which is able to conjugate with the HOMO of the benzene ring plays a dominant role in Cα-selective formation of alkenyl copper. Moreover, we also confirmed that enones were formed by C–C concerted coupling of alkenyl copper with acyl bromide instead of the oxidative addition suggested in the literature. Notably, the origin of the regioselectivity of 1,2-reduction over 1,4-reduction is mainly the steric effect, whether for the terminal alkyne or the internal alkyne case.
 Please wait while we load your content...
Please wait while we load your content...

