
A theoretical study on the electronic structure and phosphorescence properties of two series of iridium(iii) complexes with a four-membered Ir–S–C–S chelating ring†
 bc
Xiaohong
Shang
*a
bc
Xiaohong
Shang
*a
Abstract
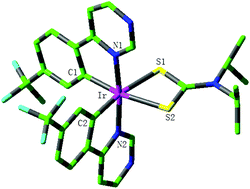
The electronic structure and photophysical properties of two series of iridium(III) complexes with a four-membered Ir–S–C–S chelating ring have been theoretically studied using the density functional theory (DFT) and time-dependent density functional theory (TDDFT) method. The calculated geometrical parameters for complexes 1c and 2c are in good agreement with the available experimental values. Furthermore, 1c and 2c possess the smallest ΔEL→H levels in the two series of complexes, respectively. The lowest lying absorption of 1c (2c) has the obvious redshift in contrast to those of 1a (2a) and 1b (2b). The lowest emission wavelengths of 1b (2b) and 1c (2c) have the obvious blue-shift and red-shift, respectively, in comparison with those of 1a (2a), which indicates that the introduction of the benzene ring into a different position in the main ligand has a different effect on the photophysical properties. The calculated results show that extending the π-conjugation of the main ligand can tune the emission of the iridium complex and has an important effect on the photophysical properties. The complex 2c possibly possesses the largest kr value among all studied complexes. It is expected that this study can be useful to understand the relationship between the structure and properties and develop efficient phosphorescent materials.
 Please wait while we load your content...
Please wait while we load your content...

