
DeepRMethylSite: a deep learning based approach for prediction of arginine methylation sites in proteins†

Abstract
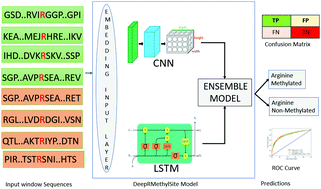
Methylation, which is one of the most prominent post-translational modifications on proteins, regulates many important cellular functions. Though several model-based methylation site predictors have been reported, all existing methods employ machine learning strategies, such as support vector machines and random forest, to predict sites of methylation based on a set of “hand-selected” features. As a consequence, the subsequent models may be biased toward one set of features. Moreover, due to the large number of features, model development can often be computationally expensive. In this paper, we propose an alternative approach based on deep learning to predict arginine methylation sites. Our model, which we termed DeepRMethylSite, is computationally less expensive than traditional feature-based methods while eliminating potential biases that can arise through features selection. Based on independent testing on our dataset, DeepRMethylSite achieved efficiency scores of 68%, 82% and 0.51 with respect to sensitivity (SN), specificity (SP) and Matthew's correlation coefficient (MCC), respectively. Importantly, in side-by-side comparisons with other state-of-the-art methylation site predictors, our method performs on par or better in all scoring metrics tested.
 Please wait while we load your content...
Please wait while we load your content...