
Photoexcited energy relaxation and vibronic couplings in π-conjugated carbon nanorings†

Abstract
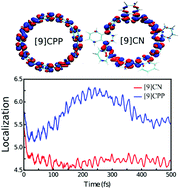
Conjugated carbon nanorings exhibit unique photophysical properties that, combined with their tunable sizes and conformations, make them suitable for a variety of practical applications. These properties are intimately associated to their strained, bent and sterically hindered cyclic structures. Herein we perform a comparative analysis of the photoinduced dynamics in carbon nanorings composed of nine phenyl units([9]CPP) and nine naphthyl units ([9]CN) respectively. The sterically demanding naphthyl units lead to large dihedral angles between neighboring units. Nevertheless, the ultrafast electronic and vibrational energy relaxation and redistribution is found to be similar for both systems. We observe that vibronic couplings, introduced by nonadiabatic energy transfer between electronic excited states, ensure the intramolecular vibrational energy redistribution through specific vibrational modes. The comparative impact of the internal conversion process on the exciton spatial localization and intra-ring migration indicates that naphthyl units in [9]CN achieve more efficient but less dynamical self-trapping compared to that of phenyl units in [9]CPP. That is, during the photoinduced process, the exciton in [9]CN is more static and localized than the exciton in [9]CPP. The internal conversion processes take place through a specific set of middle- to high-frequency normal modes, which directly influence the spatial exciton redistribution during the internal conversion, self-trapping and intra-ring migration.
- This article is part of the themed collection: 2020 PCCP HOT Articles
 Please wait while we load your content...
Please wait while we load your content...

