
First principles investigation of dissociative adsorption of H2 during CO2 hydrogenation over cubic and hexagonal In2O3 catalysts†
 ab
and
Shenggang
Li
*ac
ab
and
Shenggang
Li
*ac
Abstract
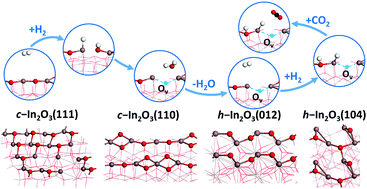
Dissociative adsorption of molecular hydrogen (H2) and migration of the adsorbed H adatom as a hydride or a proton on two of the most stable facets of the In2O3 catalyst in the cubic (c-In2O3) and hexagonal (h-In2O3) phases for CO2 hydrogenation to methanol were investigated by extensive density functional theory (DFT) calculations. Due to the relatively small variation in the oxygen vacancy (Ov) formation energy with respect to surface Ov coverage of these In2O3 surfaces, especially c-In2O3(110) and h-In2O3(012), no limit on the Ov coverage is expected to exist under the typical reaction conditions. Thus, we consider three scenarios for the dissociative adsorption of H2 and the migration of the H adsorbate, namely on the perfect (stoichiometric) In2O3 surfaces, on the partially reduced In2O3 surfaces with a single Ov site, and on the fully reduced In2O3 surfaces. Our DFT calculations show that the oppositely charged In and O pair sites on the perfect and partially reduced In2O3 surfaces facilitate the heterolytic dissociation of H2, leading to the formation of the anionic hydride at the In site crucial for CO2 hydrogenation to methanol. Our comparative studies on the four In2O3 surfaces suggest that the h-In2O3(104) surface is superior to the other three surfaces due to the facile formation of the Ov sites at low coverage and the favorable formation of the hydride adsorbate at the In site from H2 dissociation. These results indicate that this surface might be preferred for CO2 hydrogenation to methanol.
 Please wait while we load your content...
Please wait while we load your content...

