
Measuring multiple 17O–13C J-couplings in naphthalaldehydic acid: a combined solid state NMR and density functional theory approach†‡
 a
Stephen P.
Day,
a
Dinu
Iuga,
a
Jonathan R.
Yates,
b
John D.
Wallis
c
and
John V.
Hanna
*a
a
Stephen P.
Day,
a
Dinu
Iuga,
a
Jonathan R.
Yates,
b
John D.
Wallis
c
and
John V.
Hanna
*a
Abstract
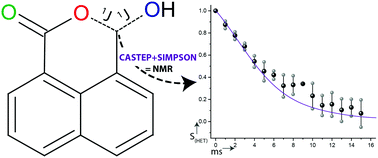
A combined multinuclear solid state NMR and gauge included projected augmented wave, density functional theory (GIPAW DFT) computational approach is evaluated to determine the four heteronuclear 1J(13C,17O) couplings in solid 17O enriched naphthalaldehydic acid. Direct multi-field 17O magic angle spinning (MAS), triple quantum MAS (3QMAS) and double rotation (DOR) experiments are initially utilised to evaluate the accuracy of the DFT approximations used in the calculation of the isotropic chemical shifts (δiso), quadrupole coupling constants (CQ) and asymmetry (ηQ) parameters. These combined approaches give δiso values of 313, 200 and 66 ppm for the carbonyl (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), ether (–O–) and hydroxyl (–OH) environments, respectively, with the corresponding measured quadrupole products (PQ) being 8.2, 9.0 and 10.6 MHz. The geometry optimised DFT structure derived using the CASTEP code gives firm agreement with the shifts observed for the ether (δiso = 223, PQ = 9.4 MHz) and hydroxyl (δiso = 62, PQ = 10.5 MHz) environments but the unoptimised experimental XRD structure has better agreement for the carbonyl group (δiso = 320, PQ = 8.3 MHz). The determined δiso and ηQ values are shown to be consistent with bond lengths closer to 1.222 Å (experimental length) rather than the geometry optimised length of 1.238 Å. The geometry optimised DFT 1J(13C,17O) coupling to the hydroxyl is calculated as 20 Hz and the couplings to the ether were calculated to be 37 (O–CO) and 32 (O–C–OH) Hz. The scalar coupling parameters for the unoptimised experimental carbonyl group predict a 1J(13C,17O) value of 28 Hz, whilst optimisation gives a value of 27 Hz. These calculated 1J(13C,17O) couplings, together with estimations of the probability of each O environment being isotopically labelled (determined by electrospray ionisation mass spectrometry) and the measured refocussable transverse dephasing (T2′) behaviour, are combined to simulate the experimental decay behaviour. Good agreement between the measured and calculated decay behaviour is observed.
O), ether (–O–) and hydroxyl (–OH) environments, respectively, with the corresponding measured quadrupole products (PQ) being 8.2, 9.0 and 10.6 MHz. The geometry optimised DFT structure derived using the CASTEP code gives firm agreement with the shifts observed for the ether (δiso = 223, PQ = 9.4 MHz) and hydroxyl (δiso = 62, PQ = 10.5 MHz) environments but the unoptimised experimental XRD structure has better agreement for the carbonyl group (δiso = 320, PQ = 8.3 MHz). The determined δiso and ηQ values are shown to be consistent with bond lengths closer to 1.222 Å (experimental length) rather than the geometry optimised length of 1.238 Å. The geometry optimised DFT 1J(13C,17O) coupling to the hydroxyl is calculated as 20 Hz and the couplings to the ether were calculated to be 37 (O–CO) and 32 (O–C–OH) Hz. The scalar coupling parameters for the unoptimised experimental carbonyl group predict a 1J(13C,17O) value of 28 Hz, whilst optimisation gives a value of 27 Hz. These calculated 1J(13C,17O) couplings, together with estimations of the probability of each O environment being isotopically labelled (determined by electrospray ionisation mass spectrometry) and the measured refocussable transverse dephasing (T2′) behaviour, are combined to simulate the experimental decay behaviour. Good agreement between the measured and calculated decay behaviour is observed.
 Please wait while we load your content...
Please wait while we load your content...

