
Combined density functional theory and molecular dynamics study of Sm0.75A0.25Co1−xMnxO2.88 (A = Ca, Sr; x = 0.125, 0.25) cathode material for next generation solid oxide fuel cell†

Abstract
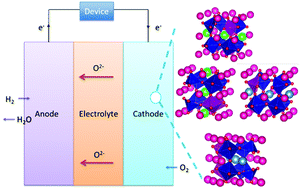
One of the main challenges facing solid oxide fuel cell (SOFC) technology is the need to develop materials capable of functioning at intermediate temperatures (500–800 °C), thereby reducing the costs associated with SOFCs. Here, Sm0.75A0.25MnxCo1−xO2.88 (A = Ca, or Sr) is investigated as a potential new cathode material to substitute the traditional lanthanum–strontium manganate for intermediate temperature SOFCs. Using a combination of density functional theory calculations and molecular dynamics simulations, the crucial parameters for SOFC performance, such as the electronic structure, electronic and ionic conductivity, and thermal expansion coefficient, were evaluated. An evaluation of the results illustrates that the conductivity and thermal match of the materials with the electrolyte is dramatically improved with respect to the existing state-of-the-art.
 Please wait while we load your content...
Please wait while we load your content...

