
Molecular dynamics based descriptors for predicting supramolecular gelation†

Abstract
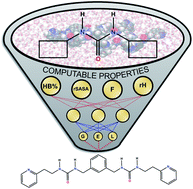
Whilst the field of supramolecular gels is rapidly moving towards complex materials and applications, their design is still an effortful and laborious trial-and-error process. Herein, we introduce four new descriptors that can be derived from all-atom molecular dynamics simulations and which are able to predict supramolecular gelation in both water and organic solvents. Their predictive ability was demonstrated via two separate machine learning techniques, a decision tree and an artificial neural network, with a dataset composed of urea-based gelators. Owing to the physical relevance of these descriptors to the supramolecular gelation process, their use could be conceptualized to other classes of supramolecular gelators and hence steer their design.
 Please wait while we load your content...
Please wait while we load your content...

