
Adsorption of amino acids on graphene: assessment of current force fields†

Abstract
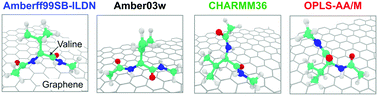
We compare the free energies of adsorption (ΔAads) and the structural preferences of amino acids on graphene obtained using the non-polarizable force fields—Amberff99SB-ILDN/TIP3P, CHARMM36/modified-TIP3P, OPLS-AA/M/TIP3P, and Amber03w/TIP4P/2005. The amino acid–graphene interactions are favorable irrespective of the force field. While the magnitudes of ΔAads differ between the force fields, the relative free energy of adsorption across amino acids is similar for the studied force fields. ΔAads positively correlates with amino acid–graphene and negatively correlates with graphene–water interaction energies. Using a combination of principal component analysis and density-based clustering technique, we grouped the structures observed in the graphene adsorbed state. The resulting population of clusters, and the conformation in each cluster indicate that the structures of the amino acid in the graphene adsorbed state vary across force fields. The differences in the conformations of amino acids are more severe in the graphene adsorbed state compared to the bulk state for all the force fields. Our findings suggest that the force fields studied will give qualitatively consistent relative strength of adsorption across proteins but different structural preferences in the graphene adsorbed state.
- This article is part of the themed collection: Soft Matter Emerging Investigators
 Please wait while we load your content...
Please wait while we load your content...

