
Using sulfur bridge oxidation to control electronic coupling and photochemistry in covalent anthracene dimers†
 b
Clàudia
Climent,
c
Peter R.
Christensen,
b
David
Casanova,
*de
Michael O.
Wolf
*b
and
Christopher J.
Bardeen
*a
b
Clàudia
Climent,
c
Peter R.
Christensen,
b
David
Casanova,
*de
Michael O.
Wolf
*b
and
Christopher J.
Bardeen
*a
Abstract
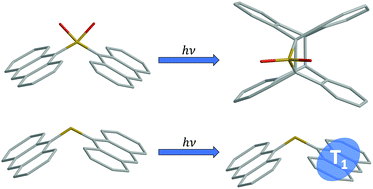
Covalently tethered bichromophores provide an ideal proving ground to develop strategies for controlling excited state behavior in chromophore assemblies. In this work, optical spectroscopy and electronic structure theory are combined to demonstrate that the oxidation state of a sulfur linker between anthracene chromophores gives control over not only the photophysics but also the photochemistry of the molecules. Altering the oxidation state of the sulfur linker does not change the geometry between chromophores, allowing electronic effects between chromophores to be isolated. Previously, we showed that excitonic states in sulfur-bridged terthiophene dimers were modulated by electronic screening of the sulfur lone pairs, but that the sulfur orbitals were not directly involved in these states. In the bridged anthracene dimers that are the subject of the current paper, the atomic orbitals of the unoxidized S linker can actively mix with the anthracene molecular orbitals to form new electronic states with enhanced charge transfer character, different excitonic coupling, and rapid (sub-nanosecond) intersystem crossing that depends on solvent polarity. However, the fully oxidized SO2 bridge restores purely through-space electronic coupling between anthracene chromophores and inhibits intersystem crossing. Photoexcitation leads to either internal conversion on a sub-20 picosecond timescale, or to the creation of a long-lived emissive state that is the likely precursor of the intramolecular [4 + 4] photodimerization. These results illustrate how chemical modification of a single atom in the covalent bridge can dramatically alter not only the photophysics but also the photochemistry of molecules.
 Please wait while we load your content...
Please wait while we load your content...

