
Computational exploration of substrate and ligand effects in nickel-catalyzed C–Si bond carboxylation with CO2†

Abstract
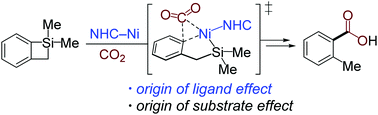
The substrate and ligand effects in nickel-catalyzed C–Si bond carboxylation with CO2 were investigated using density functional theory (DFT) calculations. Distortion/interaction analysis was used to thoroughly understand the origins of the selectivity in competing C(sp2)–Si and C(sp3)–Si bond cleavages and the reactivity of CO2 insertion when employing different cyclic organosilicon substrates and NHC ligands. The results reveal that the selectivity between C(sp2)–Si and C(sp3)–Si oxidative addition in substrates with different ring sizes is mostly determined by the interaction energy between catalysts and substrates. The deformabilities of substrates and nickelacycles are the major factors that determine the reactivity of CO2 insertion, which are derived from the substrate ring strain and the ligand steric hindrance, respectively.
 Please wait while we load your content...
Please wait while we load your content...

