
A coordination-based model for transition metal alloy nanoparticles†

Abstract
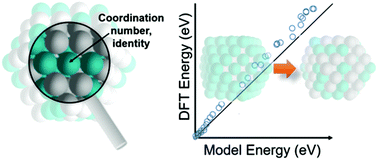
We present a simple approach for predicting the relative energies of bimetallic nanoparticles spanning a wide-ranging combinatorial space, using only the identity and nearest-neighbor coordination number of individual metal atoms as independent parameters. By performing straightforward metal atom adsorption calculations on surface slab models, we parameterize expressions for the energy of metal atoms as a function of their coordination number in 21 bimetallic pairings of fcc metals. We rigorously establish the transferability of our model by predicting relative energies of a series of nanoparticles across a large number of morphologies, sizes, atomic compositions, and arrangements. The model is particularly accurate in predicting atomic rearrangements at or near the metal surfaces, which is essential for its potential applications when studying segregation phenomena or dynamic processes in heterogeneous catalysis. By rapidly forecasting site stabilities with atomic specificity across generic structural and compositional features, our model is able to reverse engineer thermodynamically feasible motifs of active sites in bimetallic nanoparticles through robust property ⇔ structure relations.
 Please wait while we load your content...
Please wait while we load your content...

