
Redesigning hazardous chemicals by learning from structure-based drug discovery†
 *a
*a
Abstract
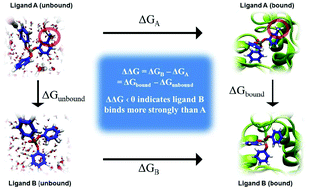
Screening all existing and new commercial chemicals using computational models is a promising alternative to economically and ethically unviable animal tests. High throughput screening (HTS) assays and quantitative structure–activity relationships (QSARs) developed to assess chemical exposure, hazard and risk are a testament to the shift toward cost-effective strategies to protect human and environmental health. While existing computational tools can be used to screen chemicals for toxic endpoints, their utility in informing design of safer chemicals is limited. Rational design of chemicals with minimal biological activity and optimal functional efficacy is the foundational principle of green chemistry; yet, it is also the least-developed field of this discipline. This paper outlines development of a computational framework for designing commercial chemicals, while considering both their biological activity and intended function. The approach resembles computational drug discovery, applied to systematically design out target-specific toxicities utilizing prior knowledge of molecular initiating events (MIE) in adverse outcomes pathways (AOP). Statistical free energy perturbation calculations are employed to demonstrate this approach for organophosphate flame retardants, a prominent class of chemicals of concern. This framework promises a strategy for safer chemical design that allows the practitioner to systematically weigh chemical drivers of toxicity, metabolism and functionality to determine trade-offs and optimal solutions to existing toxic chemicals. Complementing predictive toxicology approaches and design guidelines, this framework could advance the practice of developing safer substitutes, which is presently considered unfavourable due to high costs, prolonged development periods, and the potential of introducing a new mode of toxicity.
 Please wait while we load your content...
Please wait while we load your content...

