
Dependence of electron transfer dynamics on the number of graphene layers in π-stacked 2D materials: insights from ab initio nonadiabatic molecular dynamics†

Abstract
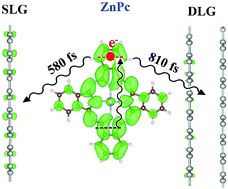
Recent time-resolved transient absorption studies demonstrated that the rate of photoinduced interfacial charge transfer (CT) from Zn-phthalocyanine (ZnPc) to single-layer graphene (SLG) is faster than to double-layer graphene (DLG), in contrast to the expectation from Fermi's golden rule. We present the first time-domain non-adiabatic molecular dynamics (NA-MD) study of the electron injection process from photoexcited ZnPc molecules into SLG and DLG substrates. Our calculations suggest that CT occurs faster in the ZnPc/SLG system than in the ZnPc/DLG system, with 580 fs and 810 fs being the fastest components of the observed CT timescales, respectively. The computed timescales are in close agreement with those reported in the experiment. The computed CT timescales are determined largely by the magnitudes of the non-adiabatic couplings (NAC), which we find to be 4 meV and 2 meV, for the ZnPc/SLG and ZnPc/DLG systems, respectively. The transitions are driven mainly by the ZnPc out-of-plane bending mode at 1100 cm−1 and an overtone of fundamental modes in graphene at 2450 cm−1. We find that dephasing occurs on the timescale of 20 fs and is similar in both systems, so decoherence does not notably change the qualitative trends in the CT timescales. We highlight the importance of proper energy level alignment for capturing the qualitative trends in the CT dynamics observed in experiment. In addition, we illustrate several methodological points that are important for accurately modeling nonadiabatic dynamics in the ZnPc/FLG systems, such as the choice of surface hopping methodology, the use of phase corrections, NAC scaling, and the inclusion of Hubbard terms in the density functional and molecular dynamics calculations.
 Please wait while we load your content...
Please wait while we load your content...

